SESSION 14. Documentation Requirements and Management for Validation. 30 March Frits Vogt
|
|
|
- Philip Page
- 6 years ago
- Views:
Transcription

1 Validation Services SESSION 14 Documentation Requirements and Management for Validation 30 March 2017 Frits Vogt


2 Content Documentation Requirements and Management for Validation I. Introduction II. Documentation Requirements III. Management for Validation IV.Interactive Exercise
3 I. Introduction Regulations & Standards Documentation Requirements Management for Validation
4 Regulation & standards EU GMP Annex 15 Qualification and Validation FDA Guidance for Industry Process Validation GHTF Study Group 3 Quality Management Systems Process Validation Guidance ISO Medical Devices - Quality Management Systems Requirements for regulatory purposes
5 EU GMP Annex DOCUMENTATION, INCLUDING VMP 2.1. Good documentation practices are important to support knowledge management throughout the product lifecycle All documents generated during qualification and validation should be approved and authorized by appropriate personnel as defined in the pharmaceutical quality system The inter-relationship between documents in complex validation projects should be clearly defined Validation protocols should be prepared which defines the critical systems, attributes and parameters and the associated acceptance criteria Qualification documents may be combined together, where appropriate, e.g. installation qualification (IQ) and operational qualification (OQ).
6 EU GMP Annex 15 (cont d) 2.6. Where validation protocols and other documentation are supplied by a third party providing validation services, appropriate personnel at the manufacturing site should confirm suitability and compliance with internal procedures before approval. Vendor protocols may be supplemented by additional documentation/test protocols before use Any significant changes to the approved protocol during execution, e.g. acceptance criteria, operating parameters etc., should be documented as a deviation and be scientifically justified Results which fail to meet the pre-defined acceptance criteria should be recorded as a deviation and be fully investigated according to local procedures. Any implications for the validation should be discussed in the report
7 EU GMP Annex 15 (cont d) 2.9. The review and conclusions of the validation should be reported and the results obtained summarized against the acceptance criteria. Any subsequent changes to acceptance criteria should be scientifically justified and a final recommendation made as to the outcome of the validation A formal release for the next stage in the qualification and validation process should be authorized by the relevant responsible personnel either as part of the validation report approval or as a separate summary document. Conditional approval to proceed to the next qualification stage can be given where certain acceptance criteria or deviations have not been fully addressed and there is a documented assessment that there is no significant impact on the next activity.
8 Control of Documents Documents required by the quality management system shall be controlled, which includes: Review, update and approval prior to issue Revision and change control Availability Prevent deterioration or loss Retention time
9 Control of Records Records are a special type of document, Additional requirements: Controls required by the quality management system must be defined, e.g. control for: Identification Storage, Security and integrity Retrieval Retention time, e.g. lifetime of drug/device + 1 year Disposition of records Protection of confidential health information Legible, readily identifiable (also after changes) and retrievable
10 Management for Validation EU GMP Annex All qualification and validation activities should be planned and take the life cycle of facilities, equipment, utilities, process and product into consideration Qualification and validation activities should only be performed by suitably trained personnel who follow approved procedures Qualification/validation personnel should report as defined in the pharmaceutical quality system although this may not necessarily be to a quality management or a quality assurance function. However, there should be appropriate quality oversight over the whole validation life cycle The key elements of the site qualification and validation programme should be clearly defined and documented in a validation master plan (VMP) or equivalent document.
11 Management for Validation EU GMP Annex The VMP or equivalent document should define the qualification/validation system 1.6. For large and complex projects, planning takes on added importance and separate validation plans may enhance clarity 1.7. A quality risk management approach should be used for qualification and validation activities. In light of increased knowledge and understanding from any changes during the project phase or during commercial production, the risk assessments should be repeated, as required. The way in which risk assessments are used to support qualification and validation activities should be clearly documented Appropriate checks should be incorporated into qualification and validation work to ensure the integrity of all data obtained.
12 II. Documentation Requirements Validation documentation Good Documentation Practices Procedures, templates, plans/protocols, reports, raw data Paper documentation / Electronic documentation Validation Archive
13 Validation Documentation The organization shall document procedures for validation of processes, including: a) defined criteria for review and approval of the processes; b) equipment qualification and qualification of personnel; c) use of specific methods, procedures and acceptance criteria; d) as appropriate, statistical techniques with rationale for sample sizes; e) requirements for records; f) revalidation, including criteria for revalidation; g) approval of changes to the processes.
14 Validation documentation The organization shall document procedures for the validation of the application of computer software used in production and service provision. Such software applications shall be validated prior to initial use and, as appropriate, after changes to such software or its application. The specific approach and activities associated with software (re)validation and shall be proportionate to the risk associated with the use of the software, including the effect on the ability of the product to conform to specifications.
15 FDA Process Validation Guidance For purposes of this guidance, process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the product and process.
16 FDA Process Validation Guidance Stage 1: Process Development Defining the commercial manufacturing process based on knowledge gained through development and scale-up activities. Stage 2: Process Qualification Confirming that the manufacturing process as designed is capable of reproducible commercial manufacturing Stage 3: Continued Process Verification Ongoing assurance is gained during routine production that the process remains in a state of control.
17 16 Process Validation (FDA) Stage 1 Process Design Product & Process Development FMEA, CPP/CQA DOE Stage 3 Continued Process Verification Process monitoring Statistical trending Process Control IPC monitoring batch records Stage 2 Process Qualification Process Performance Qualification Design Facility Qualification Utilities/Equipment
18 Validation documentation General: Each stage of Process Validation, emphasis on stage 2: Process Qualification Effective communication Transparent, comprehensive and accessible Scientific (risk based and statistically sound) Responsibilities defined, e.g. Quality Unit Deviation reporting Change control Risk control Document control
19 Validation Lifecycle documents Stage 1 Process Development Stage 2 Process Qualification Stage 3 Continued Process Verification Define Process: - URS - FS*, DS* - VMP -Traceability Matrix* Develop Process: - Risk Assessment - Engineering Study* Verify Process - FAT* - SAT* - Supplier Assessment* Qualify Process - IQ, OQ - PQ, PPQ* - VSR* Requalific ation Process Control Change Control Note: Process Development activities and documents with an * are not mandatory
20 Example of Validation documentation in a (Global) Organization Corporate procedures Divisional procedures Divisional templates Local site procedures (optional) Plans/Protocols/Reports Raw Data 19
21 Example of Validation templates and deliverables Validation of Processes Validation templates Docno. Xxx-1 Docno. Xxx-2 Docno. Xxx-3 N/A N/A Docno. Xxx-4 Docno. Xxx-5 Docno. Xxx-6 Docno. Xxx-7 Company SOP electronically available (WORD) Validation Master Plan Validation Summary Report User Requirement Specification FMEA (Equipment, Process) Traceability Matrix IQ/OQ-protocol IQ/OQ-report, including raw data PQ protocol PQ Report, including raw data
22 Good Documentation Practices Purpose of Documentation Provide detailed instructions to all personnel to insure that the product is made the same way every time. Record the exact conditions under which the product was manufactured Provide trace ability in the event of Quality problems and Recalls
23 Validation Documentation Validation provides assurance of Quality when testing 100% of the batch is not practical. Provides information that is supplied to the regulatory bodies in support of new product approval
24 Critical Concepts of Validation Documentation References Trace ability Identity of test the conditions
25 References SOP s Batch Records Drawings Analytical Methods Microbiological Methods Compendial Methods
26 Trace Ability Manufacturing System Date and Time Reagents or test materials Personnel Measuring systems Raw Materials
27 Identity of test the conditions Those parameters that define the processing conditions Time duration Temperature Pressure or Vacuum Flow Rate Agitator RPM Differential Pressure
28 How to Document? The use of data collection forms and templates During Validation errors on the side of too much information
29 Manually Recorded Data (written or using e.g. WORD) Start on a new page or form Identify what is being tested Name of the test Date and Time of test List all testing equipment All data collected Appropriate conclusions Signature at the end of the page or form
30 Electronically Recorded Data (Recorder, paper or electronic data) Identity of equipment being tested Identity of run Notebook reference if applicable Date and time Identity of recording device Traceable to the device calibration Rate at which the data was collected Signature and date Identification of various process phases
31 Supporting Documentation Batch Records Instrument records Calibration records Laboratory report forms Validation Notebooks
32 Paper Documentation Procedures for: Document Control Work flow for review and authorization Authorization Policies, Quality Manuals, Procedures, Work instructions Documents, e.g. specifications, forms, etc. Records, e.g.: BMRs Laboratory results Qualification protocols and reports 31
33 Electronic Documentation Procedures for: electronic Document Management System Work flow for review and authorization Electronic records Electronic signatures edms for the global organization Validation edms must be validated Validation procedures, work instructions Validation protocols, reports, templates Specifications, Risk Management Files, others. edms can be generic or dedicated to validation 32
34 III. Management for Validation Validation in a (Global) Organization Project management and Validation Validation in Operations Validation Master Plan Change Management and Validation Risk Management and Validation Case examples
35 Validation in a (global) organization Commitment upper management Personnel Resources Sufficient Qualified Equipment, instruments Costs Project planning Organization: Multidisciplinary: Dedicated group Subject Matter Experts R&D, engineers, production, quality
36 Project management & Validation Projects R&D project New Facility, Utility, Equipment Process IT project Changes Validation Master Plan Design & Development Quality by Design GAMP
37 Validation in Operations Roles & Responsibilities Management Production Engineers Quality Control Laboratories Quality Assurance Site Validation Master Plan Continued Process Verification Requalifications Changes
38 Validation Master Plan EU GMP Annex 15 The VMP or equivalent document should define the qualification/validation system and include or reference information on at least the following: 1. Qualification and Validation policy; 2. The organisational structure including roles and responsibilities for qualification and validation activities; 3. Summary of the facilities, equipment, systems, processes on site and the qualification and validation status; 4. Change control and deviation management for qualification and validation; 5. Guidance on developing acceptance criteria; 6. References to existing documents; 7. The qualification and validation strategy, including requalification, where applicable.
39 Change Management & Validation Change Control Procedure Change Control Request Description Impact Activities, including Validation Responsibilities Pre-approval Quality Assurance Final approval
40 New process Change Management Identify processes Change Control Low impact process High impact process Medium impact process yes Complex process Specification VMP/VSR no Risk Assessment Verification protocol/report Specifications Risk Assesment Installation Operational Qualifactions Performance Qualifactions SVMP Change Retirement
41 Risk Management & Validation Risk Management Procedure Elements of risk management process: Risk management plan Risk analysis Risk assessment Risk control Risk management report Post production and post-market surveillance information.
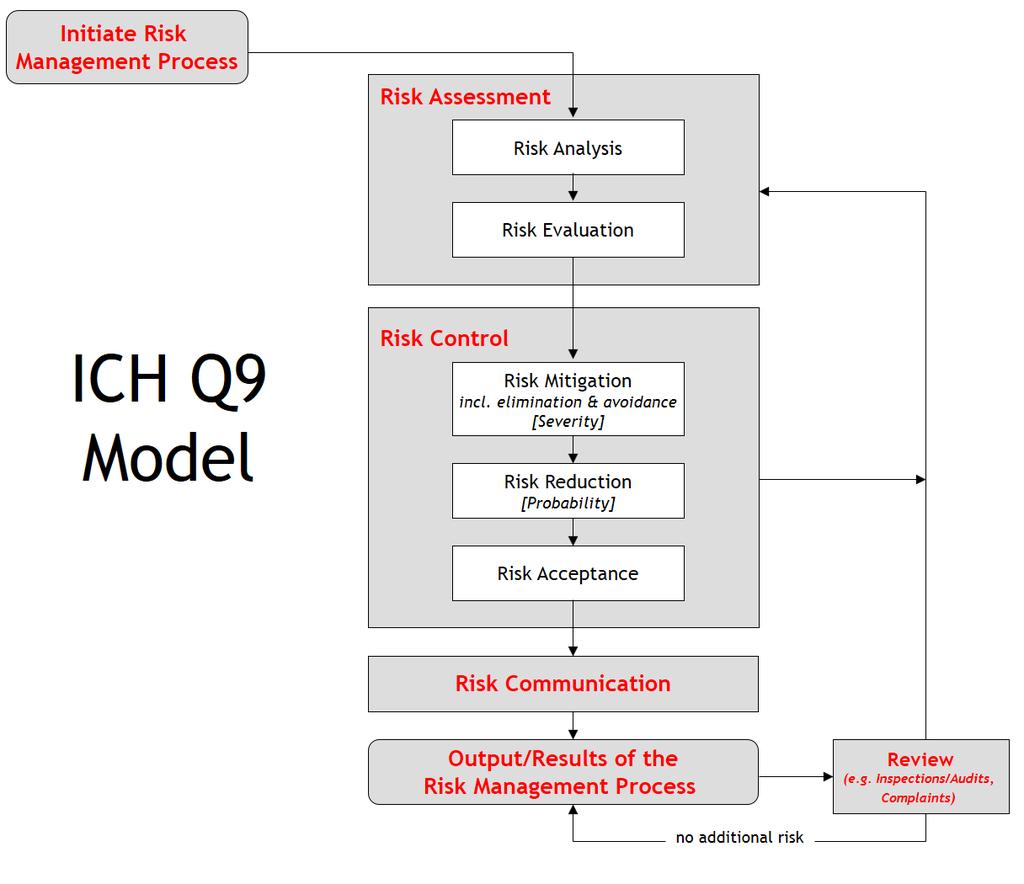
42 Risk Management
43 IV. Interactive Exercise Using real life examples for a project and regular production, participants will develop a Validation Master Plan

44 Example: new sealer PQ protocol Similar to Design Verification protocol Risk Based Testing according to SOP PQ report Similar to Design Verification protocol Risk Based Testing according to SOP
COMPLIANCE WITH EU ANNEX 15: VALIDATION AND QUALIFICATION
 COMPLIANCE WITH EU ANNEX 15: VALIDATION AND QUALIFICATION Paul L. Pluta, PhD Journal of Validation Technology Journal of GXP Compliance University of Illinois at Chicago (UIC) College of Pharmacy, Chicago,
COMPLIANCE WITH EU ANNEX 15: VALIDATION AND QUALIFICATION Paul L. Pluta, PhD Journal of Validation Technology Journal of GXP Compliance University of Illinois at Chicago (UIC) College of Pharmacy, Chicago,
MASTER PLANNING FOR VALIDATION
 MASTER PLANNING FOR VALIDATION By: Gamal Amer, Ph.D. Principal, Premier Compliance Services, Inc. 1 Process Validation: General Principles and Practices Guidance to industry issued by the FDA in January
MASTER PLANNING FOR VALIDATION By: Gamal Amer, Ph.D. Principal, Premier Compliance Services, Inc. 1 Process Validation: General Principles and Practices Guidance to industry issued by the FDA in January
EFFECTIVE MANAGEMENT OF THE PROCESS VALIDATION LIFECYCLE VALIDATION MASTER PLAN DEVELOPMENT
 EFFECTIVE MANAGEMENT OF THE PROCESS VALIDATION LIFECYCLE VALIDATION MASTER PLAN DEVELOPMENT Paul L. Pluta, PhD Journal of Validation Technology Journal of GXP Compliance University of Illinois at Chicago
EFFECTIVE MANAGEMENT OF THE PROCESS VALIDATION LIFECYCLE VALIDATION MASTER PLAN DEVELOPMENT Paul L. Pluta, PhD Journal of Validation Technology Journal of GXP Compliance University of Illinois at Chicago
CUSTOMER AND SUPPLIER ROLES AND RESPONSIBILITIES FOR 21 CFR 11 COMPLIANCE ASSESSMENT. 21 CFR Part 11 FAQ. (Frequently Asked Questions)
") 21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
PERFORMANCE QUALIFICATION PROTOCOL HVAC SYSTEM
 Page 1 of 24 PERFORMANCE QUALIFICATION PROTOCOL FOR HVAC SYSTEM Signing of this Performance Qualification Protocol indicates agreement with the Validation Master Plan approach of the equipment. Further
Page 1 of 24 PERFORMANCE QUALIFICATION PROTOCOL FOR HVAC SYSTEM Signing of this Performance Qualification Protocol indicates agreement with the Validation Master Plan approach of the equipment. Further
Annex Draft Annex
 Anmerkung 1: Dieses Vergleichsdokument wurde nach bestem Wissen und Gewissen erstellt und ist rechtlich nicht bindend. Es gelten die von der EMA veröffentlichten Dokumente in der jeweiligen Form. Anmerkung
Anmerkung 1: Dieses Vergleichsdokument wurde nach bestem Wissen und Gewissen erstellt und ist rechtlich nicht bindend. Es gelten die von der EMA veröffentlichten Dokumente in der jeweiligen Form. Anmerkung
Overview of Amgen s CQP Commissioning and Qualification Program Steve Wisniewski Principal Compliance Consultant, CAI Past Chairman ISPE C&Q COP
 May 1-3, 2012 Javits Center New York, NY Introduction to ISPE GUIDE: SCIENCE AND RISK-BASED APPROACH FOR THE DELIVERY OF FACILITIES, SYSTEMS, AND EQUIPMENT & Overview of Amgen s CQP Commissioning and Qualification
May 1-3, 2012 Javits Center New York, NY Introduction to ISPE GUIDE: SCIENCE AND RISK-BASED APPROACH FOR THE DELIVERY OF FACILITIES, SYSTEMS, AND EQUIPMENT & Overview of Amgen s CQP Commissioning and Qualification
Equipment Qualification Ensure Fit for Intended Use. IVT Amsterdam Conference Mary Sexton. 19 Mar 2015
 Equipment Qualification Ensure Fit for Intended Use IVT Amsterdam Conference Mary Sexton 19 Mar 2015 Equipment Qualification Ensure Fit for Intended Use 1. Types of Qualification 2. Validating Equipment
Equipment Qualification Ensure Fit for Intended Use IVT Amsterdam Conference Mary Sexton 19 Mar 2015 Equipment Qualification Ensure Fit for Intended Use 1. Types of Qualification 2. Validating Equipment
Risk-based Approach to Part 11 and GxP Compliance
 Welcome to our E-Seminar: Risk-based Approach to Part 11 and GxP Compliance 1 Intro Common Discussion Q: Do I really need to do this? Possible Answers A: Of course! (QA) B: Who cares, I have work to do!
Welcome to our E-Seminar: Risk-based Approach to Part 11 and GxP Compliance 1 Intro Common Discussion Q: Do I really need to do this? Possible Answers A: Of course! (QA) B: Who cares, I have work to do!
Efficient Validation Strategies and VMPs
 FDA Medical Device Industry Coalition Efficient Validation Strategies and VMPs Anne Holland Wilson Agenda History and Recent Trends Why Validate 21 CFR 820.75 Process Validation Standards and Guidance
FDA Medical Device Industry Coalition Efficient Validation Strategies and VMPs Anne Holland Wilson Agenda History and Recent Trends Why Validate 21 CFR 820.75 Process Validation Standards and Guidance
Understanding Validation Cost. Ivan Soto
 Understanding Validation Cost Ivan Soto Agenda: Understanding Validation Cost How to understand validation cost Cycle time and the impact on cost Capital versus ongoing operation cost Work force modeling
Understanding Validation Cost Ivan Soto Agenda: Understanding Validation Cost How to understand validation cost Cycle time and the impact on cost Capital versus ongoing operation cost Work force modeling
Vendor Qualification Survey
 1200 West 96 th St Minneapolis, MN 55431 Ph: 952-888-7900 Fax: 952-888-2719 Vendor Qualification Survey Vendor Information Company Name: Date: Address: City: Phone Number: email address: Product or Service
1200 West 96 th St Minneapolis, MN 55431 Ph: 952-888-7900 Fax: 952-888-2719 Vendor Qualification Survey Vendor Information Company Name: Date: Address: City: Phone Number: email address: Product or Service
White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU
 White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU Numerous articles are available comparing the current and previous EU Code of GMP Chapter 4: Documentation, but no comparison exists between
White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU Numerous articles are available comparing the current and previous EU Code of GMP Chapter 4: Documentation, but no comparison exists between
Manufacturing Optimisation Inside the Pharmaceutical Supply Chain. Business System Compliance
 Manufacturing Optimisation Inside the Pharmaceutical Supply Chain. Business System Compliance Bob Mauger, Principal Validation Consultant Cork, April 3/4 2003 Agenda Business System Compliance Overview
Manufacturing Optimisation Inside the Pharmaceutical Supply Chain. Business System Compliance Bob Mauger, Principal Validation Consultant Cork, April 3/4 2003 Agenda Business System Compliance Overview
INTERNATIONAL RESEARCH JOURNAL OF PHARMACY ISSN Review Article
 INTERNATIONAL RESEARCH JOURNAL OF PHARMACY www.irjponline.com ISSN 2230 8407 Review Article INTRODUCTION AND GENERAL OVERVIEW OF PHARMACEUTICAL PROCESS VALIDATION: A REVIEW Pandita Rachna* 1, Rana A C
INTERNATIONAL RESEARCH JOURNAL OF PHARMACY www.irjponline.com ISSN 2230 8407 Review Article INTRODUCTION AND GENERAL OVERVIEW OF PHARMACEUTICAL PROCESS VALIDATION: A REVIEW Pandita Rachna* 1, Rana A C
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL. EudraLex. The Rules Governing Medicinal Products in the European Union
 Ref. Ares(2014)968036-28/03/2014 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Medicinal products quality, safety and efficacy Brussels, 28 March 2014 EudraLex
Ref. Ares(2014)968036-28/03/2014 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Medicinal products quality, safety and efficacy Brussels, 28 March 2014 EudraLex
ICH Quality Implementation Working Group POINTS TO CONSIDER
 ICH Quality Implementation Working Group POINTS TO CONSIDER ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation Document date: 16 June 2011 International Conference on Harmonisation of Technical Requirements
ICH Quality Implementation Working Group POINTS TO CONSIDER ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation Document date: 16 June 2011 International Conference on Harmonisation of Technical Requirements
AAMI Quality Systems White Paper: Comparison of 21 CFR Part 820 to ISO 13485:2016 1
 AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
Good Automated Manufacturing Practices (GAMP)
") Good Automated Manufacturing Practices (GAMP) Klaus Krause, Amgen ISPE/GAMP Americas Steering Committee ISPE San Francisco/Bay Area Chapter Meeting, October 7, 2004 Presentation Overview I. GAMP - Organization
Good Automated Manufacturing Practices (GAMP) Klaus Krause, Amgen ISPE/GAMP Americas Steering Committee ISPE San Francisco/Bay Area Chapter Meeting, October 7, 2004 Presentation Overview I. GAMP - Organization
Guidelines for Process Validation of Pharmaceutical Dosage Forms
 Guidelines for Process Validation of Pharmaceutical Dosage Forms Version 2.1 Date issued 21/02/2010 Date of implementation 21/05/2010 31 August 2010 Page 1 of 20 Guidelines for Process Validation of Pharmaceutical
Guidelines for Process Validation of Pharmaceutical Dosage Forms Version 2.1 Date issued 21/02/2010 Date of implementation 21/05/2010 31 August 2010 Page 1 of 20 Guidelines for Process Validation of Pharmaceutical
The long anticipated draft of the FDA s
 This article provides an overview of the draft guidance, the key changes in relation to the 1987 guidance, and reviews its potential impact on the current industry approaches to science- and risk-based
This article provides an overview of the draft guidance, the key changes in relation to the 1987 guidance, and reviews its potential impact on the current industry approaches to science- and risk-based
Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)
![Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)](/thumbs/72/66853561.jpg "Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)") Regulatory Overview Annex 11 and Part 11 Sion Wyn Conformity +[44] (0) 1492 642622 sion.wyn@conform-it.com 1 Two Key Regulations Annex 11 21 CFR Part 11 Apply to the regulated company, but often have a
Regulatory Overview Annex 11 and Part 11 Sion Wyn Conformity +[44] (0) 1492 642622 sion.wyn@conform-it.com 1 Two Key Regulations Annex 11 21 CFR Part 11 Apply to the regulated company, but often have a
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C.
 Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Manufacturing Breakout Session 1 Survey Results: Drug Product Manufacturing
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Manufacturing Breakout Session 1 Survey Results: Drug Product Manufacturing
Quality Management System MANUAL. SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702
 Quality Management System MANUAL SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702 Doc. No. G5500 Rev. 11 Status : APPROVED Effective: 09/07/2017 Page 2 of 32 Quality Manual Table of Contents
Quality Management System MANUAL SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702 Doc. No. G5500 Rev. 11 Status : APPROVED Effective: 09/07/2017 Page 2 of 32 Quality Manual Table of Contents
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL. EudraLex The Rules Governing Medicinal Products in the European Union
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, 25 October 2005 EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 EU
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, 25 October 2005 EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 EU
Process Validation Guidelines. Report highlights 23 rd February 2018
 Process Validation Guidelines Report highlights 23 rd February 2018 Process validation is an important element of pharmaceutical quality system An effective system provides assurance of the continued capability
Process Validation Guidelines Report highlights 23 rd February 2018 Process validation is an important element of pharmaceutical quality system An effective system provides assurance of the continued capability
Quality Agreement. by and between. Supplier Name. Address: and. Client Name: Address:
 NOTE TO USERS This Quality Agreement template was developed by the Bulk Pharmaceutical Task Force (BPTF), an affiliate organization of the Society of Chemical Manufacturers and Affiliates (SOCMA), as a
NOTE TO USERS This Quality Agreement template was developed by the Bulk Pharmaceutical Task Force (BPTF), an affiliate organization of the Society of Chemical Manufacturers and Affiliates (SOCMA), as a
Current Features of USFDA and EMA Process Validation Guidance
 Human Journals Review Article April 2016 Vol.:6, Issue:1 All rights are reserved by Patwekar S.L et al. Current Features of USFDA and EMA Process Validation Guidance Keywords: Pharmaceutical validation,
Human Journals Review Article April 2016 Vol.:6, Issue:1 All rights are reserved by Patwekar S.L et al. Current Features of USFDA and EMA Process Validation Guidance Keywords: Pharmaceutical validation,
Validation. An Easy to Understand Guide Validating Purified Water Systems
 Validation Overview Validation Documentation Validation Activities Validation Mistakes Validation Protocols Test Selection Common Verifications When to test Test Layout and Content Validation Protocol
Validation Overview Validation Documentation Validation Activities Validation Mistakes Validation Protocols Test Selection Common Verifications When to test Test Layout and Content Validation Protocol
QbD Concepts Applied to Qualification and Transfer of Analytical Methods
 QbD Concepts Applied to Qualification and Transfer of Analytical Methods CMC Strategy Forum Latin America - 2014 Patrick Swann Senior Director Technical Development QbD = Quality by Design QbD - A systematic
QbD Concepts Applied to Qualification and Transfer of Analytical Methods CMC Strategy Forum Latin America - 2014 Patrick Swann Senior Director Technical Development QbD = Quality by Design QbD - A systematic
Initiation of Validation
 Initiation of Validation Design Review Who Validates? What Process to Validate? Process Validation Decision Tree When to Validate? Prospective Validation Concurrent Validation Retrospective Validation
Initiation of Validation Design Review Who Validates? What Process to Validate? Process Validation Decision Tree When to Validate? Prospective Validation Concurrent Validation Retrospective Validation
Reviewers, approvers and executers of this plan are captured in the approval routing tab of this document in IFS.
 DOCUMENT NUMBER: 1034722 LOCATION: FT COLLINS/LOVELAND PAGE 1 of 8 1.0 PURPOSE 1.1 The purpose of this Validation Master Plan (VMP) is to identify the validation and testing requirements necessary to qualify
DOCUMENT NUMBER: 1034722 LOCATION: FT COLLINS/LOVELAND PAGE 1 of 8 1.0 PURPOSE 1.1 The purpose of this Validation Master Plan (VMP) is to identify the validation and testing requirements necessary to qualify
Introduction to 21 CFR 11 - Good Electronic Records Management
 INTERNATIONAL JOURNAL OF ADVANCES IN PHARMACY, BIOLOGY AND CHEMISTRY Review Article Introduction to 21 CFR 11 - Good Electronic Records Management Pal Tapas Kumar* and Maity Subhasis NSHM Knowledge Campus,
INTERNATIONAL JOURNAL OF ADVANCES IN PHARMACY, BIOLOGY AND CHEMISTRY Review Article Introduction to 21 CFR 11 - Good Electronic Records Management Pal Tapas Kumar* and Maity Subhasis NSHM Knowledge Campus,
GMP On Site Series. GMP Essentials
 GMP On Site Series GMP Essentials GMP Basics Objectives 1. State the critical definitions of the pharmaceutical industry. 2. Describe the law as it applies to various critical functions. 3. State the history
GMP On Site Series GMP Essentials GMP Basics Objectives 1. State the critical definitions of the pharmaceutical industry. 2. Describe the law as it applies to various critical functions. 3. State the history
Lifecycle Management of Process Analytical Technology Procedures
 Lifecycle Management of Process Analytical Technology Procedures IFPAC 2015 Marta Lichtig Senior Scientist in New Testing Technologies, ACS Member Contents General Comparison : PV guide to NIR model development
Lifecycle Management of Process Analytical Technology Procedures IFPAC 2015 Marta Lichtig Senior Scientist in New Testing Technologies, ACS Member Contents General Comparison : PV guide to NIR model development
HISTORY AND MILESTONES
 HISTORY AND MILESTONES HISTORY AND MILESTONES DOC S.r.l., Documentation Organization & Consultancy, was established in 1997 by MASCO group C.E.O. Eng. Alberto Borella and Eng. Paolo Curtò who became Managing
HISTORY AND MILESTONES HISTORY AND MILESTONES DOC S.r.l., Documentation Organization & Consultancy, was established in 1997 by MASCO group C.E.O. Eng. Alberto Borella and Eng. Paolo Curtò who became Managing
Computerised Systems. Alfred Hunt Inspector. Wholesale Distribution Information Day, 28 th September Date Insert on Master Slide.
 Computerised Systems Wholesale Distribution Information Day, Alfred Hunt Inspector Date Insert on Master Slide Slide 1 Index What is a computerised system Updates to EU GDPs Expectations Case studies Slide
Computerised Systems Wholesale Distribution Information Day, Alfred Hunt Inspector Date Insert on Master Slide Slide 1 Index What is a computerised system Updates to EU GDPs Expectations Case studies Slide
Computerised System Validation
 Computerised System Validation Considerations for the Validation Lifecycle Paul Moody GMP Conference 12 th November 2014 Regulatory References EU GMP Annex 11 (2011) http://ec.europa.eu/health/files/eudralex/vol-4/annex11_01-2011_en.pdf
Computerised System Validation Considerations for the Validation Lifecycle Paul Moody GMP Conference 12 th November 2014 Regulatory References EU GMP Annex 11 (2011) http://ec.europa.eu/health/files/eudralex/vol-4/annex11_01-2011_en.pdf
Process validation in medical devices
 Process validation in medical devices Fulfil requirements with expert regulatory guidance 1TÜV SÜD Contents INTRODUCTION 4 VALIDATION PLANNING 5 INSTALLATION QUALIFICATION 7 OPERATIONAL QUALIFICATION 9
Process validation in medical devices Fulfil requirements with expert regulatory guidance 1TÜV SÜD Contents INTRODUCTION 4 VALIDATION PLANNING 5 INSTALLATION QUALIFICATION 7 OPERATIONAL QUALIFICATION 9
Quality Risk Management (ICH Q9): WHO Model for Sustainable Quality Medicines
: WHO Model for Sustainable Quality Medicines") Quality Risk Management (ICH Q9): WHO Model for Sustainable Quality Medicines Longe Sunday Anthony Head- Quality Assurance May & Baker Nigeria Plc. Pharmacentre, Ota, Nigeria eaglesconsult@gmail.com; Slonge@may-
Quality Risk Management (ICH Q9): WHO Model for Sustainable Quality Medicines Longe Sunday Anthony Head- Quality Assurance May & Baker Nigeria Plc. Pharmacentre, Ota, Nigeria eaglesconsult@gmail.com; Slonge@may-
References Concept. Principle. EU Annex 11 US FDA , (g), (i), 11 Orlando Lopez 2/15/11. Old Annex 11.
, (i), 11 Orlando Lopez 2/15/11. Old Annex 11.") References Concept Principle a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which
References Concept Principle a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which
Jean Guichard. Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN
 #: Internet-WP Version: e1.00 /EN") Jean Guichard White Paper Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN Introduction to Good Process Record Management (GxP) 2 / 6 Document History Version Date
Jean Guichard White Paper Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN Introduction to Good Process Record Management (GxP) 2 / 6 Document History Version Date
Guidelines for validation and qualification, including change control, for hospital transfusion laboratories
 Transfusion Medicine GUIDELINES Guidelines for validation and qualification, including change control, for hospital transfusion laboratories British Committee for Standards in Haematology, Transfusion
Transfusion Medicine GUIDELINES Guidelines for validation and qualification, including change control, for hospital transfusion laboratories British Committee for Standards in Haematology, Transfusion
CSV Inspection Readiness through Effective Document Control. Eileen Cortes April 27, 2017
 CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
Validation Strategies for Equipment from Multiple Vendors
 Welcome to our E-Seminar: Validation Strategies for Equipment from Multiple Vendors Page 1 Content FDA guidelines and inspectional observations Validation & Qualification Approaches Instrument Qualification
Welcome to our E-Seminar: Validation Strategies for Equipment from Multiple Vendors Page 1 Content FDA guidelines and inspectional observations Validation & Qualification Approaches Instrument Qualification
Seamless Integration of ASTM E2500, Annex 15, FDA Process Validation Guideline and Chinese GMP in Large CapEx Project in China Daniel Nilsson
 Seamless Integration of ASTM E2500, Annex 15, FDA Process Validation Guideline and Chinese GMP in Large CapEx Project in China Daniel Nilsson Senior Management Consultant Agenda Introduction A state-of
Seamless Integration of ASTM E2500, Annex 15, FDA Process Validation Guideline and Chinese GMP in Large CapEx Project in China Daniel Nilsson Senior Management Consultant Agenda Introduction A state-of
FDA Process Validation
 Page 1 of 5 Published on Controlled Environments Magazine (http://www.cemag.us) Home > FDA Process Validation FDA Process Validation Wai Wong Working with the New 2011 Guidelines. In January 2011, the
Page 1 of 5 Published on Controlled Environments Magazine (http://www.cemag.us) Home > FDA Process Validation FDA Process Validation Wai Wong Working with the New 2011 Guidelines. In January 2011, the
Guidance for Industry Process Validation: General Principles and Practices. F. Hoffman-La Roche, Ltd.
 Guidance for Industry Process Validation: General Principles and Practices D M k T k Dr. Mark Tucker, F. Hoffman-La Roche, Ltd. Disclosures I am currently a Senior Technical Advisor at F. Hoffman -La Roche.
Guidance for Industry Process Validation: General Principles and Practices D M k T k Dr. Mark Tucker, F. Hoffman-La Roche, Ltd. Disclosures I am currently a Senior Technical Advisor at F. Hoffman -La Roche.
OMCL Network of the Council of Europe QUALITY ASSURANCE DOCUMENT
 OMCL Network of the Council of Europe QUALITY ASSURANCE DOCUMENT PA/PH/OMCL (13) 113 2R Evaluation and Reporting of Results Core document Full document title and reference Evaluation and Reporting of Results
OMCL Network of the Council of Europe QUALITY ASSURANCE DOCUMENT PA/PH/OMCL (13) 113 2R Evaluation and Reporting of Results Core document Full document title and reference Evaluation and Reporting of Results
Correspondence Between ISO 13485:2016 and 21 CFR Part 820 QMS Requirements
 Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
WHO PUBLIC INSPECTION REPORT (WHOPIR) Finished Product Manufacturer. Mylan Nashik ( Sinnar in CRM) AND
 Finished Product Manufacturer. Mylan Nashik ( Sinnar in CRM) AND") SOP 408.4 Annex D 20, avenue Appia CH-1211 Geneva 27 Switzerland Tel central +41 22 791 2111 Fax central +41 22 791 3111 www.who.int WHO PUBLIC INSPECTION REPORT (WHOPIR) Finished Product Manufacturer
SOP 408.4 Annex D 20, avenue Appia CH-1211 Geneva 27 Switzerland Tel central +41 22 791 2111 Fax central +41 22 791 3111 www.who.int WHO PUBLIC INSPECTION REPORT (WHOPIR) Finished Product Manufacturer
Validation and Automated Validation
 TOP INDUSTRY QUESTIONS Validation and Automated Validation 1 Table of Contents 03 04 07 10 13 16 19 INTRODUCTION SECTION 1 - Validation Standards How is validation defined under Title 21 CFR Part 11? What
TOP INDUSTRY QUESTIONS Validation and Automated Validation 1 Table of Contents 03 04 07 10 13 16 19 INTRODUCTION SECTION 1 - Validation Standards How is validation defined under Title 21 CFR Part 11? What
PMDA Perspective: Regulatory Updates on Process Validation Standard
 CMC Strategy Forum Japan 2014 Tokyo, Japan, December 8-9, 2014 PMDA Perspective: Regulatory Updates on Validation Standard Kazunobu Oyama, PhD Office of Cellular and Tissue-based Products PMDA, Japan Disclaimer:
CMC Strategy Forum Japan 2014 Tokyo, Japan, December 8-9, 2014 PMDA Perspective: Regulatory Updates on Validation Standard Kazunobu Oyama, PhD Office of Cellular and Tissue-based Products PMDA, Japan Disclaimer:
QUALITY MANUAL. This document defines the requirements, processes, structure and documentation for the Teledyne DGO Quality Management System.
 Page 1 of 13 1. PURPOSE This document defines the requirements, processes, structure and documentation for the Teledyne DGO Quality Management System. 2. SCOPE The Teledyne DGO Quality Management System
Page 1 of 13 1. PURPOSE This document defines the requirements, processes, structure and documentation for the Teledyne DGO Quality Management System. 2. SCOPE The Teledyne DGO Quality Management System
á1058ñ ANALYTICAL INSTRUMENT QUALIFICATION
 First Supplement to USP 40 NF 35 General Information / á1058ñ Analytical Instrument Qualification 1 á1058ñ ANALYTICAL INSTRUMENT QUALIFICATION INTRODUCTION A large variety of analytical instruments, ranging
First Supplement to USP 40 NF 35 General Information / á1058ñ Analytical Instrument Qualification 1 á1058ñ ANALYTICAL INSTRUMENT QUALIFICATION INTRODUCTION A large variety of analytical instruments, ranging
ZOLL Document Number: 90E0004 Page 5 of 15 Maintenance of Quality Records
 ZOLL Number: 90E0004 Page 5 of 15 1. INTRODUCTION 1.1 Purpose 1.2 Scope The purpose of this document is to provide guidance regarding the identification of a quality record and the documentation practices
ZOLL Number: 90E0004 Page 5 of 15 1. INTRODUCTION 1.1 Purpose 1.2 Scope The purpose of this document is to provide guidance regarding the identification of a quality record and the documentation practices
PROPOSAL FOR REVISION OF THE SUPPLEMENTARY GUIDELINES ON GOOD MANUFACTURING PRACTICES: VALIDATION, APPENDIX 7:NON-STERILE PROCESS VALIDATION
 1 Working document QAS/13.527/Rev.2 August 2014 RESTRICTED 2 3 4 5 6 7 8 9 10 PROPOSAL FOR REVISION OF THE SUPPLEMENTARY GUIDELINES ON GOOD MANUFACTURING PRACTICES: VALIDATION, APPENDIX 7:NON-STERILE PROCESS
1 Working document QAS/13.527/Rev.2 August 2014 RESTRICTED 2 3 4 5 6 7 8 9 10 PROPOSAL FOR REVISION OF THE SUPPLEMENTARY GUIDELINES ON GOOD MANUFACTURING PRACTICES: VALIDATION, APPENDIX 7:NON-STERILE PROCESS
EU Annex 11 US FDA 211, 820, 11; other guidelines Orlando López 11-MAY-2011
 Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
CALIBRATION AND QUALIFICATION OF EQUIPMENTS IN THE PHARMACEUTICAL INDUSTRY: EMPHASIS ON RADIOPHARMACEUTICALS PRODUCTION
 2011 International Nuclear Atlantic Conference - INAC 2011 Belo Horizonte, MG, Brazil, October 24-28, 2011 ASSOCIAÇÃO BRASILEIRA DE ENERGIA NUCLEAR - ABEN ISBN: 978-85-99141-04-5 CALIBRATION AND QUALIFICATION
2011 International Nuclear Atlantic Conference - INAC 2011 Belo Horizonte, MG, Brazil, October 24-28, 2011 ASSOCIAÇÃO BRASILEIRA DE ENERGIA NUCLEAR - ABEN ISBN: 978-85-99141-04-5 CALIBRATION AND QUALIFICATION
INS QA Programme Requirements
 Specification Date: 20/3/17 INS QA Programme Requirements UNCONTROLLED WHEN PRINTED Author: J Cooch AUTHORISATION Date: 20/3/17 A Brown Owner: J Cooch (Signature) N.B. only required for hard copy If issued
Specification Date: 20/3/17 INS QA Programme Requirements UNCONTROLLED WHEN PRINTED Author: J Cooch AUTHORISATION Date: 20/3/17 A Brown Owner: J Cooch (Signature) N.B. only required for hard copy If issued
Health Products and Food Branch Inspectorate
 Our Mandate: To promote good nutrition and informed use of drugs, food, medical devices and natural health products, and to maximize the safety and efficacy of drugs, food, natural health products, medical
Our Mandate: To promote good nutrition and informed use of drugs, food, medical devices and natural health products, and to maximize the safety and efficacy of drugs, food, natural health products, medical
GMP In-house Training
 Your benefits: Customised to fit your company s requirements - cost-effective and flexible! GMP In-house Training for the Pharmaceutical, API and Medical Device Industry We offer practice-oriented GMP
Your benefits: Customised to fit your company s requirements - cost-effective and flexible! GMP In-house Training for the Pharmaceutical, API and Medical Device Industry We offer practice-oriented GMP
FO-5 PR-1, FO-1,2 PR-1 EM-6 PR-1, FE-1, NA-1 FO-10 FO-7, EM-2 EM-2. ISO Environmental Management Systems - Specification Yes.
 ISO 14001 Environmental Management Systems - Specification Yes Minor No Comments/Questions 4.1 GENERAL The organization shall establish and maintain an environmental management system, the requirements
ISO 14001 Environmental Management Systems - Specification Yes Minor No Comments/Questions 4.1 GENERAL The organization shall establish and maintain an environmental management system, the requirements
A COMPARATIVE FRAMEWORK BETWEEN NEW PRODUCT & LEGACY PRODUCT PROCESS VALIDATION
 A COMPARATIVE FRAMEWORK BETWEEN NEW PRODUCT & LEGACY PRODUCT PROCESS VALIDATION By Mark Mitchell, Principal Consultant, Process and Engineering, Pharmatech Associates, Inc. PHARMATECH WHITE PAPER.DOCX
A COMPARATIVE FRAMEWORK BETWEEN NEW PRODUCT & LEGACY PRODUCT PROCESS VALIDATION By Mark Mitchell, Principal Consultant, Process and Engineering, Pharmatech Associates, Inc. PHARMATECH WHITE PAPER.DOCX
QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES
 Your Company Name QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES Origination Date: XXXX Document Identifier: Date: Document Revision: QMS-00 QMS Policies and Procedures Latest Revision Date Abstract:
Your Company Name QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES Origination Date: XXXX Document Identifier: Date: Document Revision: QMS-00 QMS Policies and Procedures Latest Revision Date Abstract:
Library Guide: Active Pharmaceutical
 Library Guide: Active Pharmaceutical Ingredients (API) Table of Contents Overview...3 Sample Curriculum...5 Course Descriptions: A Step-by-Step Approach to Process Validation (PHDV79)...7 A Tour of the
Library Guide: Active Pharmaceutical Ingredients (API) Table of Contents Overview...3 Sample Curriculum...5 Course Descriptions: A Step-by-Step Approach to Process Validation (PHDV79)...7 A Tour of the
Prequalification Team WHO PUBLIC INSPECTION REPORT Vaccine Manufacturer
 Part 1: General information Name of Manufacturer Production Block Physical address Contact address Prequalification Team WHO PUBLIC INSPECTION REPORT Vaccine Manufacturer. Clean Utilities in the Basement.
Part 1: General information Name of Manufacturer Production Block Physical address Contact address Prequalification Team WHO PUBLIC INSPECTION REPORT Vaccine Manufacturer. Clean Utilities in the Basement.
HACCP audit checklist
 Requirement HACCP audit checklist Prerequisite Program Management Commitment 1. Senior management ensures that the responsibilities and authorities are defined and communicated within the company Internal
Requirement HACCP audit checklist Prerequisite Program Management Commitment 1. Senior management ensures that the responsibilities and authorities are defined and communicated within the company Internal
The Skyworks Quality Management System strives to:
 Skyworks has embraced a workplace where Quality is the number one differentiator to achieve Customer Loyalty. Skyworks has adopted a single Quality Management System which drives efficiency, consistency
Skyworks has embraced a workplace where Quality is the number one differentiator to achieve Customer Loyalty. Skyworks has adopted a single Quality Management System which drives efficiency, consistency
The validation of biological Active Pharmaceutical
 This article presents a procedure for the validation of biological API manufacturing processes. It details each step of the procedure for biological processes applications, and covers the regulatory requirements
This article presents a procedure for the validation of biological API manufacturing processes. It details each step of the procedure for biological processes applications, and covers the regulatory requirements
Strategies for Risk Based Validation of Laboratory Systems
 Strategies for Risk Based Validation of Laboratory Systems Video Web Seminar September 23, 2004 Ludwig Huber E-mail: ludwig_huber@agilent.com Today s Agenda Background information: why risk assessment,
Strategies for Risk Based Validation of Laboratory Systems Video Web Seminar September 23, 2004 Ludwig Huber E-mail: ludwig_huber@agilent.com Today s Agenda Background information: why risk assessment,
How to support compliance with GAMP 5
 PRESENTATION How to support compliance with GAMP 5 KERESZTESI Kálmán Controsys Irányítástechnikai Kft. kalman.keresztesi@controsys.hu Agenda What is GAMP 5 about? Application Lifecycle Management in automation
PRESENTATION How to support compliance with GAMP 5 KERESZTESI Kálmán Controsys Irányítástechnikai Kft. kalman.keresztesi@controsys.hu Agenda What is GAMP 5 about? Application Lifecycle Management in automation
COMPUTERISED SYSTEMS
 ANNEX 11 COMPUTERISED SYSTEMS PRINCIPLE This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components
ANNEX 11 COMPUTERISED SYSTEMS PRINCIPLE This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components
Implementation Life-cycle SOP
 1.0 Commercial in Confidence 11-Aug-2006 1 of 12 Implementation Life-cycle SOP Document No: H4_SOP_0100 Prepared by: David Brown Date: 11-Aug-2006 Version: 1.0 1.0 Commercial in Confidence 11-Aug-2006
1.0 Commercial in Confidence 11-Aug-2006 1 of 12 Implementation Life-cycle SOP Document No: H4_SOP_0100 Prepared by: David Brown Date: 11-Aug-2006 Version: 1.0 1.0 Commercial in Confidence 11-Aug-2006
Quality Manual. Index
 This quality policy manual is for the use of, its Clients, Vendors, and the appropriate Regulatory Agencies. Unauthorized duplication or transfer to others is prohibited. Index Authorization... 3 Introduction...
This quality policy manual is for the use of, its Clients, Vendors, and the appropriate Regulatory Agencies. Unauthorized duplication or transfer to others is prohibited. Index Authorization... 3 Introduction...
MASTER VALIDATION PLAN PROCEDURE DRAFT DRAFT OF A POSSIBLE COMPUTER SYSTEMS VALIDATION MASTER PLAN PROCEDURE
 DRAFT OF A POSSIBLE COMPUTER SYSTEMS VALIDATION MASTER PLAN PROCEDURE 1. PURPOSE The purpose of this SOP is to define the minimum requirements for Computer Systems Validation (CSV) at (insert name of company
DRAFT OF A POSSIBLE COMPUTER SYSTEMS VALIDATION MASTER PLAN PROCEDURE 1. PURPOSE The purpose of this SOP is to define the minimum requirements for Computer Systems Validation (CSV) at (insert name of company
HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL. ISO 22000:2005 Quality Manual
 HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL ISO 22000:2005 Quality Manual QUALITY MANUAL ISO 22000:2005 Food Safety Management HACCPEuropa Publications 2012 Table of Contents Introduction...
HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL ISO 22000:2005 Quality Manual QUALITY MANUAL ISO 22000:2005 Food Safety Management HACCPEuropa Publications 2012 Table of Contents Introduction...
System Requirements (URS)
") System s (URS) System Document Control System Document ID URS000XXX Version 1 DOCUMENT APPROVALS Reason For Signature Name Position Signature Date Prepared by Vic Johnson Project Manager Checked for accuracy
System s (URS) System Document Control System Document ID URS000XXX Version 1 DOCUMENT APPROVALS Reason For Signature Name Position Signature Date Prepared by Vic Johnson Project Manager Checked for accuracy
Conducting Supplier Audits: Ensure Validation Compliance
 Conducting Supplier Audits: Ensure Validation Compliance Presented by: Gamal Amer, Ph. D. Principal Premier Compliance Services, Inc. All rights reserved. Do not copy without permission. 1 Outline Why
Conducting Supplier Audits: Ensure Validation Compliance Presented by: Gamal Amer, Ph. D. Principal Premier Compliance Services, Inc. All rights reserved. Do not copy without permission. 1 Outline Why
For analytical laboratories. A primer Good laboratory practice and current good manufacturing practice
 For analytical laboratories A primer Good laboratory practice and current good manufacturing practice For analytical laboratories A primer Good laboratory practice and current good manufacturing practice
For analytical laboratories A primer Good laboratory practice and current good manufacturing practice For analytical laboratories A primer Good laboratory practice and current good manufacturing practice
ISO 9001: 2000 (December 13, 2000) QUALITY MANAGEMENT SYSTEM DOCUMENTATION OVERVIEW MATRIX
 QUALITY MANAGEMENT SYSTEM DOCUMENTATION OVERVIEW MATRIX") In completing your Documented Quality Management System Review, it is important that the following matrix be completed and returned to us as soon as possible. This will save time during the review and
In completing your Documented Quality Management System Review, it is important that the following matrix be completed and returned to us as soon as possible. This will save time during the review and
Quality Assessment & GMP Similarities & Differences
 Quality Assessment & GMP Similarities & Differences EMEA, Monday 26 th October 2009 Cormac Dalton Inspector Irish Medicines Board Date 12-Oct-09 Slide 1 Content Overview of commonalities & differences
Quality Assessment & GMP Similarities & Differences EMEA, Monday 26 th October 2009 Cormac Dalton Inspector Irish Medicines Board Date 12-Oct-09 Slide 1 Content Overview of commonalities & differences
22 January Division of Dockets Management (HFA 305) Food and Drug Administration 5630 Fishers Lane, rm Rockville, MD 20852
 Food and Drug Administration 5630 Fishers Lane, rm Rockville, MD 20852") 22 January 2009 Division of Dockets Management (HFA 305) Food and Drug Administration 5630 Fishers Lane, rm. 1061 Rockville, MD 20852 SUBMISSION OF COMMENTS, DOCKET NO. FDA-2008-D-0559 Dear Sir or Madam:
22 January 2009 Division of Dockets Management (HFA 305) Food and Drug Administration 5630 Fishers Lane, rm. 1061 Rockville, MD 20852 SUBMISSION OF COMMENTS, DOCKET NO. FDA-2008-D-0559 Dear Sir or Madam:
35 STATION DOUBLE ROTORY COMPRESSION MACHINE
 Page 1 of 16 User Requirement Specifications 35 STATION DOUBLE ROTORY Equipment ID: T-35COMP 01 Page 2 of 16 Table of Contents 1.0 APPROVAL SIGNATURES 2.0 OVERVIEW 3.0 PROCESS DESCRIPTION 4.0 PRODUCTIVITY
Page 1 of 16 User Requirement Specifications 35 STATION DOUBLE ROTORY Equipment ID: T-35COMP 01 Page 2 of 16 Table of Contents 1.0 APPROVAL SIGNATURES 2.0 OVERVIEW 3.0 PROCESS DESCRIPTION 4.0 PRODUCTIVITY
At Your ServIce. Microbiology Services. The life science business of Merck operates as MilliporeSigma in the U.S. and Canada.
 At Your ServIce Microbiology Services The life science business of Merck operates as MilliporeSigma in the U.S. and Canada. Microbiology Services Optimize your QC lab workflow and ensure regulatory compliance
At Your ServIce Microbiology Services The life science business of Merck operates as MilliporeSigma in the U.S. and Canada. Microbiology Services Optimize your QC lab workflow and ensure regulatory compliance
Regulatory Expectations, Standards & Guidelines
 Regulatory Expectations, Standards & Guidelines Regulatory Requirements Pharmacopeias Good Automated Manufacturing Practice (GAMP) 21 CFR Part 11 and Annex 11 Consequences of Non-Compliance 22 Regulatory
Regulatory Expectations, Standards & Guidelines Regulatory Requirements Pharmacopeias Good Automated Manufacturing Practice (GAMP) 21 CFR Part 11 and Annex 11 Consequences of Non-Compliance 22 Regulatory
SYSTEMKARAN ADVISER & INFORMATION CENTER QUALITY MANAGEMENT SYSTEM ISO9001:
 SYSTEM KARAN ADVISER & INFORMATION CENTER QUALITY MANAGEMENT SYSTEM ISO9001:2015 WWW.SYSTEMKARAN.ORG 1 WWW.SYSTEMKARAN.ORG Foreword... 5 Introduction... 6 0.1 General... 6 0.2 Quality management principles...
SYSTEM KARAN ADVISER & INFORMATION CENTER QUALITY MANAGEMENT SYSTEM ISO9001:2015 WWW.SYSTEMKARAN.ORG 1 WWW.SYSTEMKARAN.ORG Foreword... 5 Introduction... 6 0.1 General... 6 0.2 Quality management principles...
GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED GXP ENVIRONMENTS
 PHARMACEUTICAL INSPECTION CONVENTION PHARMACEUTICAL INSPECTION CO-OPERATION SCHEME PI 011-2 1 July 2004 PIC/S GUIDANCE GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED GXP ENVIRONMENTS PIC/S July 2004
PHARMACEUTICAL INSPECTION CONVENTION PHARMACEUTICAL INSPECTION CO-OPERATION SCHEME PI 011-2 1 July 2004 PIC/S GUIDANCE GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED GXP ENVIRONMENTS PIC/S July 2004
Choosing the Right Instrument Vendor for Complete, Cost-Effective Spectrometer Compliance
 White Paper Choosing the Right Instrument Vendor for Complete, Cost-Effective Spectrometer Compliance Key Words 21 CFR Part 11 cgmp Compliance GLP ISO Molecular Spectroscopy Qualification Six Sigma Validation
White Paper Choosing the Right Instrument Vendor for Complete, Cost-Effective Spectrometer Compliance Key Words 21 CFR Part 11 cgmp Compliance GLP ISO Molecular Spectroscopy Qualification Six Sigma Validation
WHO Prequalification of In Vitro Diagnostics Programme
 P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Manufacturing Site(s) Inspection (Assessment of the Quality Management System) WHO Prequalification
P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Manufacturing Site(s) Inspection (Assessment of the Quality Management System) WHO Prequalification
Risk-Based Approach to Software Quality and Validation
 Risk-Based Approach to Software Quality and Validation By Praxis Life Sciences 1925 West Field Court, Suite 125, Lake Forest, IL 60045 praxislifesciences.com +1(847) 295-7160 validationcenter.com Risk
Risk-Based Approach to Software Quality and Validation By Praxis Life Sciences 1925 West Field Court, Suite 125, Lake Forest, IL 60045 praxislifesciences.com +1(847) 295-7160 validationcenter.com Risk
DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS
 GLOBAL PROVIDER, LOCAL SOLUTIONS IN YOUR LANGUAGE DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS Francesco Amorosi PhD Octorber 2017 DATA INTEGRITY & CSV:
GLOBAL PROVIDER, LOCAL SOLUTIONS IN YOUR LANGUAGE DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS Francesco Amorosi PhD Octorber 2017 DATA INTEGRITY & CSV:
Version 1.0 August 30, 2017
 Supplier Requirements Guideline 1.0 Purpose The purpose of this Supplier Requirements Guideline is to provide a guidance document with the expectations and requirements Baxter has of its suppliers. The
Supplier Requirements Guideline 1.0 Purpose The purpose of this Supplier Requirements Guideline is to provide a guidance document with the expectations and requirements Baxter has of its suppliers. The
Good Documentation Practices. Documentation Requirements For Regulated Environments
 Good Documentation Practices Documentation Requirements For Regulated Environments Agenda Example Deficiency 2014 Result Of Poor Record Creation Data Integrity EU GMP Chapter 4 Requirements Non GMP Compliant
Good Documentation Practices Documentation Requirements For Regulated Environments Agenda Example Deficiency 2014 Result Of Poor Record Creation Data Integrity EU GMP Chapter 4 Requirements Non GMP Compliant
SQF 2000 Code. 6th Edition AUGUST A HACCP-Based Supplier Assurance Code for the Food Manufacturing and Distributing Industries
 SQF 2000 Code A HACCP-Based Supplier Assurance Code for the Food Manufacturing and Distributing Industries 6th Edition AUGUST 2008 Safe Quality Food Institute 2345 Crystal Drive, Suite 800 Arlington, VA
SQF 2000 Code A HACCP-Based Supplier Assurance Code for the Food Manufacturing and Distributing Industries 6th Edition AUGUST 2008 Safe Quality Food Institute 2345 Crystal Drive, Suite 800 Arlington, VA
SIMATIC IT. SIMATIC IT R&D SUITE V7.1 Compliance Response ERES. Introduction 1. The Requirements in Short 2
 Introduction 1 The Requirements in Short 2 SIMATIC IT Meeting the Requirements with SIMATIC IT R&D Suite V7.1 3 SIMATIC IT R&D SUITE V7.1 Evaluation List for SIMATIC IT R&D Suite V7.1 4 Product Information
Introduction 1 The Requirements in Short 2 SIMATIC IT Meeting the Requirements with SIMATIC IT R&D Suite V7.1 3 SIMATIC IT R&D SUITE V7.1 Evaluation List for SIMATIC IT R&D Suite V7.1 4 Product Information
TECTURA LIFE SCIENCES HIGHLIGHTS
 HIGHLIGHTS BATCH AND SERIAL NUMBER MANAGEMENT QUALITY CONTROL VENDOR RATING ELECTRONIC SIGNATURE AND AUDIT TRAIL R&D MODULE MANAGEMENT OF ACTIVE INGREDIENTS SEQUENCE PLANNING MOBILE SCANNER INTEGRATION
HIGHLIGHTS BATCH AND SERIAL NUMBER MANAGEMENT QUALITY CONTROL VENDOR RATING ELECTRONIC SIGNATURE AND AUDIT TRAIL R&D MODULE MANAGEMENT OF ACTIVE INGREDIENTS SEQUENCE PLANNING MOBILE SCANNER INTEGRATION
ISO Part 1 and Part 2 Compliance Requirements. Cathriona O Neill
 ISO 11607 Part 1 and Part 2 Compliance Requirements Cathriona O Neill I.S. EN 11607 Introduction ISO 11607 is the principal guidance document. Packaging for terminally sterilised medical devices - Part
ISO 11607 Part 1 and Part 2 Compliance Requirements Cathriona O Neill I.S. EN 11607 Introduction ISO 11607 is the principal guidance document. Packaging for terminally sterilised medical devices - Part
QUALITY MANUAL. Number: M-001 Revision: C Page 1 of 18 THIS DOCUMENT IS CONSIDERED UNCONTROLLED UNLESS ISSUED IDENTIFIED AS CONTROLLED
 Page 1 of 18 THIS DOCUMENT IS CONSIDERED UNCONTROLLED UNLESS ISSUED IDENTIFIED AS CONTROLLED Page 2 of 18 REVISION HISTORY DATE CHANGE DESCRIPTION 10/11/06 Original release 10/21/09 Revised to ISO9001:2008
Page 1 of 18 THIS DOCUMENT IS CONSIDERED UNCONTROLLED UNLESS ISSUED IDENTIFIED AS CONTROLLED Page 2 of 18 REVISION HISTORY DATE CHANGE DESCRIPTION 10/11/06 Original release 10/21/09 Revised to ISO9001:2008
Stanley Industries, Inc. ISO 9001:2008 Quality Policy Manual
 Stanley ISO 9001:2008 Table of Contents and STANLEY Document Reference Related STANLEY Section Page Procedure(s) 1. Introduction 1 None 2. Scope 1 None 3. Organizational Structure & 1 STANLEY Company History
Stanley ISO 9001:2008 Table of Contents and STANLEY Document Reference Related STANLEY Section Page Procedure(s) 1. Introduction 1 None 2. Scope 1 None 3. Organizational Structure & 1 STANLEY Company History