Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome
|
|
|
- Tracey Parsons
- 6 years ago
- Views:
Transcription
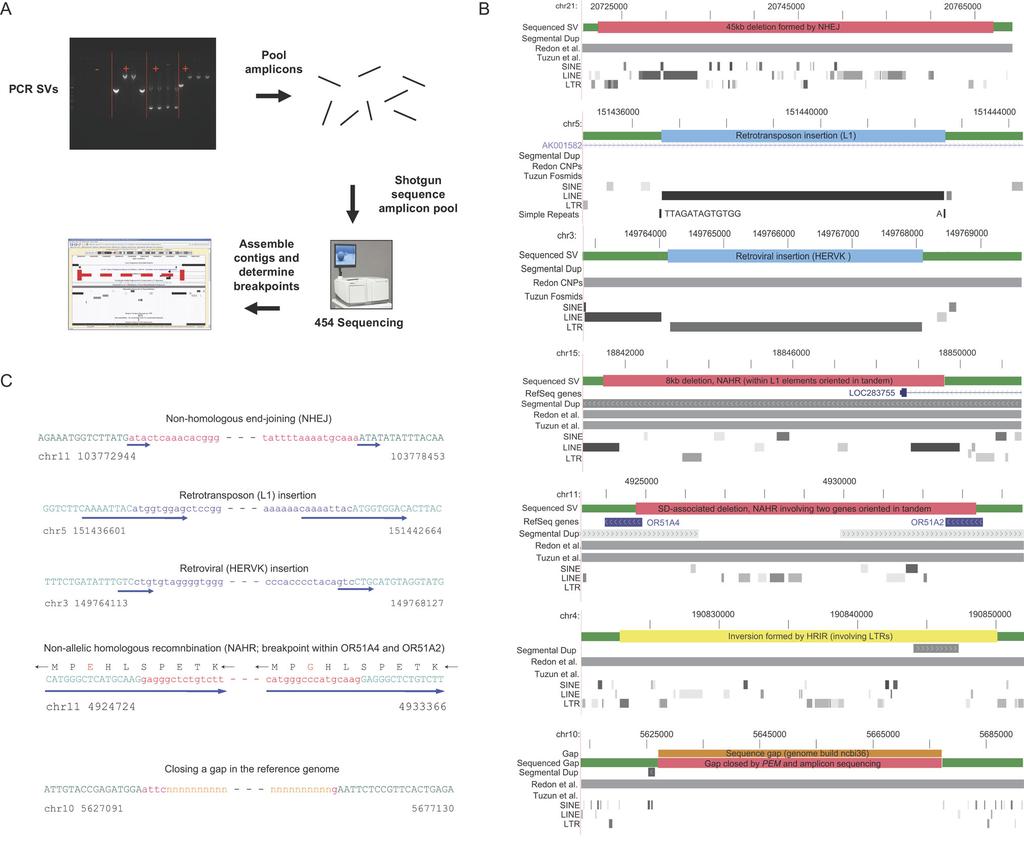
1 Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome Jan O. Korbel, 1,2 * Alexander Eckehart Urban, 3 * Jason P. Affourtit, 4 * Brian Godwin, 4 Fabian Grubert, 5 Jan Fredrik Simons, 4 Philip M. Kim, 1 Dean Palejev, 5 Nicholas J. Carriero, 6 Lei Du, 4 Bruce E. Taillon, 4 Zhoutao Chen, 4 Andrea Tanzer, 7,8,9 A. C. Eugenia Saunders, 3 Jianxiang Chi, 10 Fengtang Yang, 10 Nigel P. Carter, 10 Matthew E. Hurles, 10 Sherman M. Weissman, 5 Timothy T. Harkins, 11 Mark B. Gerstein, 1,6,12 Michael Egholm, 4 Michael Snyder 1,3 1 Molecular Biophysics and Biochemistry Department, Yale University, New Haven, CT 06520, USA. 2 European Molecular Biology Laboratory, Heidelberg, Germany. 3 Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06520, USA Life Sciences, A Roche Company, Branford, CT 06405, USA. 5 Department of Genetics, Yale University School of Medicine, New Haven, CT 06520, USA. 6 Department of Computer Science, Yale University, New Haven, CT 06520, USA. 7 Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT 06520, USA. 8 Department of Computer Science, University of Leipzig, Leipzig, Germany. 9 Institute for Theoretical Chemistry, University of Vienna, 1090 Vienna, Austria. 10 The Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK. 11 Roche Applied Science, Indianapolis, IN 46250, USA. 12 Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA. *These authors contributed equally to this work. To whom correspondence should be addressed. megholm@454.com (M.E.) or michael.snyder@yale.edu (M.S.) Structural variation of the genome involves kilobase- to megabase-sized deletions, duplications, insertions, inversions, and complex combinations of rearrangements. We introduce high-throughput and massive paired-end mapping (PEM), a large-scale genome sequencing method to identify structural variants (SVs) ~3 kb or larger that combines the rescue and capture of paired-ends of 3-kb fragments, massive 454 Sequencing, and a computational approach to map DNA reads onto a reference genome. PEM was used to map SVs in an African and putatively European individual and identified shared and divergent SVs relative to the reference genome. Overall, we finemapped more than 1000 SVs and documented that the number of SVs among humans is much larger than initially hypothesized; many of the SVs potentially affect gene function. The breakpoint junction sequences of more than 200 SVs were determined with a novel pooling strategy and computational analysis. Our analysis provided insights into the mechanisms of SV formation in humans. Structural variation of large segments (>50kb) of the human genome was recently found to be widespread in healthy individuals (1-4), with ~4000 affected genomic loci currently listed in the Database of Genomic Variants (DGV; (2)). Structural variants (SVs) may have a more significant impact on phenotypic variation than Single Nucleotide Polymorphisms (SNPs) (4, 5). SVs have been implicated in gene expression variation (5), female fertility (6), susceptibility to HIV infection (7), systemic autoimmunity (8), and genomic disorders such as Williams-Beuren Syndrome and Velocardiofacial Syndrome (9, 10). Thus, understanding the full extent of structural variation is important for understanding phenotypic variation and genetic disease in humans. Previous methods for detecting SVs used comparative genome hybridization (array-cgh, which involves DNA microarrays and detects copy-number variants, or CNVs; (4)), and fosmid paired end sequencing (FPES; (3)) at relatively low resolution (>50 kb for array-cgh, >8kb for FPES). Importantly, these methods map SVs below the resolution where breakpoints can be detected (for array-cgh), or are laborious (for FPES). Consequently, breakpoint junction sequences of a limited number of SVs/CNVs have been reported (2, 3, 11). Methods for comprehensively detecting SVs <10 kb, which may encompass most variants, and for mapping breakpoints, are lacking; thus, how SVs affect genes and the mechanism(s) by which SVs form are not known. Development of paired-end mapping for detecting SVs. In order to identify SVs more accurately we developed Paired-End Mapping (PEM), which involves the preparation and isolation of paired-ends of 3kb fragments (12), and their massive sequencing with 454 technology (Fig. 1) (13). The large number of paired-end reads were optimally mapped to the human genome computationally (12). Structural rearrangements were identified as significant differences between the fragments identified by the paired-end reads and the corresponding regions of the reference sequence. Five different signatures (i-v) were used to predict SVs (12) (Fig. 1B). (i) Deletions relative to the reference genome were / / 13 September 2007 / Page 1 / /science
2 identified by paired-ends spanning a genomic region in the reference genome longer than a specified cutoff (Fig. 1). (ii) Simple insertions relative to the reference genome were predicted with paired-ends that spanned a region shorter than a cutoff. (iii) Mated insertions contained sequences connected to a distal locus on the basis of their paired-ends. (iv) Inversions were detected through a relative orientation different from the reference genome. (v) Unmated insertions contained sequences connected to a distal locus; one of the two expected breakpoints remained undetected. Unless stated otherwise, we treated insertions and deletions as SV indels because a deletion in one individual is synonymous to an insertion in the other. These events can be distinguished with additional analyses (see below). For all rearrangement types (i)-(v), we required that SVs were supported by at least two independent paired-end reads to eliminate false positives that may arise from rare chimerical constructs that can form during the ligation reaction (12). This approach identifies deletions, inversions, mated insertions, and unmated insertions that are ~3 kb or larger, and simple insertions 2-3 kb in size. From two or more paired-end sequences per SV, we obtained an average breakpoint resolution of 644 bp (12), a range that facilitates the validation of SVs by PCR. PEM detection of SVs in the human genome. We applied PEM to map SVs in the genomes of two individuals: a female (NA15510) in which 297 SV events had been mapped with FPES (12), and a second female (NA18505; Yoruba, Ibadan, from Nigeria) previously analyzed in the international HapMap project (14). The ancestry of NA15510 is unknown, however, the individual appears to be of European descent as described below. We sequenced over 10 million (NA15510) and 21 million (NA18505) paired-ends yielding effective coverages of 2.1- and 4.3-fold relative to the 6 billion base-pair diploid genome (12) to identify ~62% and 93% of the SVs, respectively (12). We identified 1175 SV indels (853 deletions, 322 insertions, i.e. 39 simple, 82 mated, 201 unmated) and 122 inversions, for a total of 1297 SV events (Table 1 and tables S1, S2). For 20% of these events, only one out of two expected breakpoint junctions were identified (particularly in the European sample, which lacks saturation). Extrapolating to full coverage, we predict 761 and 887 SV events relative to the reference genome for NA15510 and NA18505, respectively, at this level of resolution. SVs were distributed throughout the genome with a number of hotspots (Fig. 2), such as an 8 Mb region at 22q11.2 containing 13 SVs, and a 18 Mb region at 7q11 containing 29 SVs. Both regions are involved in relatively frequent genomic disorders (Velocardiofacial Syndrome, and Williams-Beuren Syndrome, respectively), and SVs in healthy individuals at those loci was previously observed at lower resolution (e.g. (2)). We compared the SVs identified in NA15510 to NA18505, and found that nearly half (45%) of the predicted SVs were shared between them (table S3): i.e., 43% of the deletions, 52% of the insertions, and 43% of the inversions (12). Thus, a considerable fraction of the SV events occur commonly in the population and are presumably ancient. It is also possible that common SVs are due to errors in the human reference sequence. However, this is likely to be rare as 18 of 19 cases we tested by PCR contained the reference sequence in one or more DNA samples. Thus, many of the detected events are bona fide SVs and likely to commonly occur in humans. We were able to confirm 41% of all deletion and inversion events predicted in (3) for NA Since only 62% of NA15510 is covered in our study, extrapolation to full coverage predicts that PEM would identify ~65% of all SVs predicted in (3), including 70% of the deletions. False positives may account for some of the discrepancies between studies, although 83% and 97% of predicted events were confirmed by (3) and us respectively (see below). It is also possible that these two studies have different, conservative thresholds [see (3) and (12)], reducing the identification of true events. Regardless, PEM identified an additional 407 SVs (377 SV indels, 30 inversions) in NA15510 not previously detected, including many events <8kb and also larger variants. Similarly, the number of SVs detected in NA18505 is higher than those previously identified at lower resolution (4), with an additional 813 SVs identified and finemapped. The majority of SVs detected by PEM were small (Fig. 3). ~65% of all SVs were <10 kb and 30% were <5kb, however, 15% of all predicted SVs were larger than 100 kb and events up to Mb-level in size were predicted; size distributions were similar for NA15510 and NA In addition, the size and extent of SVs found indicates that healthy individuals differ by several megabases of nucleotide sequence (Fig. 3B, table S1). We analyzed the fraction of heterozygous and homozygous SVs by PCR analysis (for both NA15510 and NA18505) and we searched for the allele represented in the human reference genome with paired-end sequences (for NA18505 (12)). Our results confirmed a previous study (3) and revealed that 23% and 15 20% of the SVs in NA15510 and NA18505, respectively, are homozygous (12). SV validation. To validate PEM-SVs, PCR analysis was performed on 40 randomly chosen samples with 5 sets of primers spanning predicted breakpoint junctions (12). Of 34 SVs that could be scored, 33 (97%) yielded a single, clear PCR band at the expected size range (12). SVs were also confirmed and validated with five additional approaches: (i) comparison with SVs in DGV (2), (ii) comparison with an alternative human genome assembly ( Celera assembly ), (iii) DNA microarray-based high-resolution comparative genome / / 13 September 2007 / Page 2 / /science
3 hybridization (array-cgh) (15, 16), (iv) fiber-based fluorescent-in situ hybridization (fiber-fish) (v) and a onepass PCR assay spanning SV breakpoint junctions. We found that 59% and 60%, respectively, of NA15510 and NA18505 SVs intersected with SVs represented in DGV (Table 1); the figures increase to 91% and 90%, respectively, for variants in the range kb (12). Since the resolution of most SVs in DGV is low [>50kb; (17)], it is unclear whether the overlapping variants correspond to the same event. Comparison with the Celera assembly confirmed 104 (22%) and 103 (12%) of the NA15510 and NA18505 SVs, respectively (12). The fraction of events shared with the Celera assembly is probably higher, as many (>200) SV regions aligned poorly or coincided with gaps in the Celera assembly and were thus excluded from this analysis (12). The observation that a higher fraction of SVs is shared between NA15510 and the Celera assembly (which is primarily derived from a donor of European ancestry) indicates that NA15510 is of European origin. Array-CGH experiments compared NA15510 DNA to NA18505 DNA with a set of eight oligonucleotide tiling arrays covering non-repetitive regions of the genome. Of 48 NA15510-specific indels represented by at least 10 probes on the array [our detection limit (12)], 31 (65%) were validated by array-cgh (see Fig. 4; table S1). The imperfect overlap may be because either some NA15510 SVs intersect with SVs in NA18505 and thus may not yield good array signals and/or array-cgh misses a portion of true positive events (4). For four inversions, not reported previously in DGV (2), we performed fiber-fish on stretched DNA and located PEM-identified inversion breakpoints at the correct position for three (Fig. 4; table S1). We were unable to detect the fourth inversion, presumably because its size ( 4 kb) is below our detection limit for fiber-fish. In order to validate SVs for downstream sequence analyses, we further analyzed 261 SVs predicted in NA15510, and 616 predicted in NA18505 in a one-pass PCR test, focusing primarily on SVs not represented in the Celera assembly. For 249 SVs DNA from a total of four individuals was analyzed (Fig. 4): NA15510, NA18505, NA11997 (European ancestry), and NA18614 (Asian). 58% of the predicted SVs were validated by PCR in one or more individuals, including the sample in which the SV was originally identified. For the tests performed on all four individuals, 89% shared SVs among two or more individuals and 48% shared SVs in all four individuals. We also examined segregation patterns of 5 SVs in parent-offspring trios and observed Mendelian segregation patterns of SVs in 9 meioses (12). Thus, their presence in multiple individuals and segregation patterns indicate that the majority of SVs are genetically stable and unlikely to have formed de novo, or in the cultured cells that were analyzed (4). Altogether, 551 unique SVs were validated by array-cgh, fiber-fish, the Celera assembly, and/or PCR (table S1). Overlap of SVs with genes and functional elements. We compared the locations of predicted SVs smaller than 100 kb (which are of high confidence (12)) with annotated genes and functional elements. We found that many (17%) of the SVs in both individuals may directly affect gene function by removing exons or fusing annotated genes (40 RefSeq genes), lying in introns (243 genes), or altering the copy-number or orientation of protein coding genes (32 genes). The fraction of SVs affecting genes is slightly less than that expected by chance (Fig. 3C), suggesting selective constraint against SVs (4). We also analyzed protein coding genes by their Gene Ontology (GO) functional classes. Consistent with previous observations (1-4), we found genes involved in organismal physiological processes (e.g., immunity, and cell-cell signaling; P<1e-14; hypergeometric test; Bonferroni correction) to be enriched with SVs (12), whereas genes involved in cellular physiological processes were depleted (P<0.001; Fig. 3D). Genes encoding proteins involved in interactions with the environment such as immune response, perception of smell, and perception of chemical stimuli were particularly likely to harbor SVs (12). Retrovirus and transposition related proteins also contain more SVs than expected by chance. Genome-wide analysis of SVs and associated breakpoints. To study SV formation, we determined the sequences surrounding breakpoint junctions with a new high throughput approach (12). PCR products containing breakpoints were pooled, sequenced with 454 technology (13), and contigs assembled (Fig. 5A). Breakpoints for a nonredundant set of 114 SVs were deduced with either a high-quality contig or at least two separate 454 reads (table S1). This method was most successful for SVs with breakpoints in regions that either have non-identical DNA sequences or share short (<200 bp) identity at the junctions. The sequence data also allowed us to identify 344 putative SNPs located adjacent to the sequenced SVs (12), which may serve as useful future predictors for the SVs (table S4). The 114 sequenced SVs included events confirmed by the Celera assembly. Manual inspection of sequence alignments in 14 cases indicated that all 14 correspond to the same SVs evident in the Celera assembly (12). We therefore included in our analyses an additional 88 (non-redundant) SVs confirmed by the 'Celera assembly' for which breakpoints could be assigned at high confidence, yielding a total of 202 SVs identified by PEM with sequenced breakpoint junctions (188 SV Indels, and 14 inversions). The types of events observed from sequenced SVs were similar to those deduced from the Celera assembly. We initially examined the association of breakpoint junctions with elements in the human genome. Several / / 13 September 2007 / Page 3 / /science
4 studies (e.g. reviewed in (10)) have suggested an association of SVs with segmental duplications (SDs); following the analysis scheme in (18) we find that 28 of 202 SVs have at least one breakpoint that directly intersects a SD (~2.6-fold enrichment over the genomic background, P< from permutations (12)). Furthermore, many SVs occurred in short to medium-sized repetitive elements (30 for Alu/SINE elements, 74 for L1/LINEs, 3 for L2/LINEs, and 30 for LTRs). Out of the latter, L1 elements are significantly enriched (with P<0.01), whereas L2 elements appear significantly depleted (P<0.0001). Finally, Alu elements are not significantly enriched near SVs, despite previous reports (19). Mechanisms of SV formation and effects on genes. Detailed manual analysis of the breakpoint junctions of SV indels revealed likely mechanisms as to how most SVs arose (see Fig. 5, B and C) and in most cases allowed us to distinguish insertion and deletion events. For example, entire LINE elements with polya tails near the breakpoint junctions are inferred to be insertion events; recombination between homologous regions resulting in sequence loss indicates deletions. Insertion and deletion events can be further confirmed by comparison with other primate sequences (12). Most SV indels originated from nonhomologous end joining (NHEJ; 56%) and retrotransposition events (30%). NHEJ (20), in which breakpoint junctions were flanked by nonhomologous regions (except for short stretches of duplicated sequence ('microhomology', typically <5bp, that immediately flank the junction), was prevalent even among large SVs (Fig. 4A, and table S1) and in regions with large SDs. Most (90%) retrotransposition events were due to LINEs although a small fraction (8%) corresponded to SVA elements (21). We also observed one instance of retroposition by an Endogenous Retrovirus despite conflicting reports suggesting that these are not active or move infrequently in humans (21). Our finding indicates that these elements have been mobile in relatively recent human history (22). DNA transposition events (21) were not observed. SVs have been found to be associated with duplicated regions suggesting that many form by nonallelic homologous recombination (NAHR). Even though SVs and SDs are strongly associated, relatively few events (14% of all SV indels) are likely mediated by NAHR (defined as homologous regions flanking the breakpoint junctions (12)). NAHR was rare even for large SVs as only 2 of 21 SV indels >20kb in size originated from NAHR. 18 were formed through NHEJ, and for one the mechanism was unassigned. NAHR events were located in: (i) highly repetitive elements LINE/L1 elements (4 cases), LTR elements (5 cases), SINEs (6 cases) and simple sequences (2 cases), and (ii) high complexity regions, SDs (5 cases) and unique DNA (5 cases). As an interesting example of the latter, we observed a fusion involving the protein coding regions of two olfactory receptor (OR) genes, OR51A4 and OR51A2 resulting in a new gene predicted to encode a protein identical to OR51A4, with upstream regions from OR51A2 (Fig. 5, B and C). OR51A4 and OR51A2 are present in the rhesus monkey confirming that the ancestral region contains both genes and that SVformation involved a recent gene-fusion event. We suggest that deviation in gene content for the large OR gene family may lead to diversity of olfactory perception in the human population. In addition to NHEJ, retrotransposition, and NAHR, other events may have occurred or could not be assigned. In 4 cases, simple sequence DNA was present at the breakpoint junctions; NAHR or other mechanisms may be involved in their formation (23). 4 cases were unassigned and two sequenced SVs closed gaps in the human reference sequence (see e.g. Fig. 5, B and C). We also analyzed 14 inversions. 4 instances of homologous recombination between inverted repeats (HRIR) were observed; surprisingly, the remaining 10 inversions appeared to involve events that do not require homology. Overall, a large fraction of all of the SVs we sequenced (at least 57%) had one or both breakpoints in non-repetitive sequence, indicating that high-complexity genomic regions are subject to variation. Discussion. PEM enabled global detection of SVs at 3 kb resolution, and an average resolution of breakpoint assignment of 644 bp. We identified approximately 1300 SVs in two individuals, suggesting that humans may differ to a greater extent in SVs/CNVs than SNPs, when considering the total number of nucleotides affected. To date most human genome sequencing projects do not directly analyze SVs. Our study reveals that given their high frequency it will be essential to incorporate SV detection into human genome sequencing projects (24). Overall, PEM is a cost effective method both for improving genome assemblies and for revealing SVs present in the genome to ultimately understand human diversity. PEM has several advantages over existing methods. First, PEM increases resolution of SV detection to the level of confirmation by PCR, and resolution can be further improved by more careful selection of evenly sized DNA fragments for circularization. Second, PEM does not require preparation of a DNA library that involves cloning. However, the short size of fragments (3 kb) used in this study hampers the detection of simple insertions >3kb, although larger insertions can be detected by their mated ends. Similar to other SV detection methods a limitation of PEM is that SVs in regions with multiple copies of highly similar and long (>3 kb) repeats are difficult to identify. Fortunately, although 45% of the human genome is comprised of high copy-number repeat elements, these are often sufficiently divergent or short, and can thus be / / 13 September 2007 / Page 4 / /science
5 distinguished by PEM. Additional refinements of PEM are also possible, and will eventually allow detection of all SVs in the human genome. References and Notes 1. J. Sebat et al., Science 305, 525 (Jul 23, 2004). 2. A. J. Iafrate et al., Nat Genet 36, 949 (Sep, 2004). 3. E. Tuzun et al., Nat Genet 37, 727 (Jul, 2005). 4. R. Redon et al., Nature 444, 444 (Nov 23, 2006). 5. B. E. Stranger et al., Science 315, 848 (Feb 9, 2007). 6. H. Stefansson et al., Nat Genet 37, 129 (Feb, 2005). 7. E. Gonzalez et al., Science 307, 1434 (Mar 4, 2005). 8. M. Fanciulli et al., Nat Genet 39, 721 (May 21, 2007). 9. J. R. Lupski, P. Stankiewicz, PLoS Genet 1, e49 (Dec, 2005). 10. J. L. Freeman et al., Genome Res 16, 949 (Aug, 2006). 11. J. O. Korbel et al., Proc Natl Acad Sci U S A 104, (June 5, 2007). 12. Approaches are described in detail in the Supplementary Online Materials and Methods section. 13. M. Margulies et al., Nature 437, 376 (Sep 15, 2005). 14. D. Altshuler et al., Nature 437, 1299 (Oct 27, 2005). 15. R. R. Selzer et al., Genes Chromosomes Cancer 44, 305 (Nov, 2005). 16. A. E. Urban et al., Proc Natl Acad Sci U S A 103, 4534 (Mar 21, 2006). 17. B. P. Coe et al., Genomics 89, 647 (May, 2007). 18. P. M. Kim et al., in prepartion (available at J. A. Bailey, G. Liu, E. E. Eichler, Am J Hum Genet 73, 823 (Oct, 2003). 20. E. V. Linardopoulou et al., Nature 437, 94 (Sep 1, 2005). 21. R. E. Mills, E. A. Bennett, R. C. Iskow, S. E. Devine, Trends Genet 23, 183 (Apr, 2007). 22. R. Belshaw et al., J Virol 79, (Oct, 2005). 23. A. Bacolla, R. D. Wells, J Biol Chem 279, (Nov 12, 2004). 24. R. Khaja et al., Nat Genet 38, 1413 (Nov 22, 2006). 25.We thank C. Turcotte, C. Celone, D. Riches, and 454 colleagues, and R. Bjornson at the Yale High Performance Computation Center (funded by NIH grant: RR ) for technical support. Funding was provided by a Marie Curie Fellowship (JOK), the Alexander von Humboldt Foundation (AT), the Wellcome Trust (NPC, MEH, JC and FY), Roche Applied Science, and the NIH (Yale CEGS grant). Accessions can be found in table S5, and at Supporting Online Material Materials and Methods Tables S1to S6 Fig. S1 References 21 August 2007; accepted 13 September 2007 Published online 27 September 2007; /science Include this information when citing this paper. Fig. 1. Paired-end mapping (PEM). (A) Flow chart illustrating PEM: (i) Genomic DNA was sheared to yield DNA fragments ~3kb; (ii) biotinylated hairpin adapters were ligated to the fragment ends; (iii) fragments were circularized (iv) and randomly sheared; (v) linker (+) fragments were isolated; (vi) the library was subjected to 454 Sequencing (13). (vii) Paired-ends were analyzed computationally to determine (viii) the distribution of paired-end spans (shown for a single 454 Sequencing pool). (B) Types of SVs. Deletions were predicted from paired-end spans larger than a specified cutoff D; simple insertions: span < cutoff I; inversions: ends map to the genome at different relative orientations; other types of insertions (defined in the text as mated and unmated) were detected with evidence of sequence integration from a distal locus. Fig. 2. SVs identified in two humans. (A) SVs mapped onto chromosomal ideograms (12). Right side: Red=deletion; blue=insertion; yellow=inversion; double length indicates SVs observed in both individuals. Left side: log-scale size of an event (events 1 Mb are drawn at same length, corresponding to the right-side line); unmated insertions (i.e. events lacking a predicted breakpoint and thus size information (12)) and simple insertions (12) are depicted with 1 kb lines; line colors indicate repetitive sequences in ±3kb window of the predicted breakpoint junction (12): red=sds; blue=lines; yellow=ltrs; green=satellites; black=two or more repetitive elements with equal frequency; gray=no repeat association. The arrow indicates the region in B. A high-resolution image of this figure is available as fig. S1. (B) Amplified view of chromosome 4 region. SVs in NA18505 are indicated with dashed lines (validation: squares); NA15510, dotted lines (validation: circle). SVs shared between individuals: solid line. Colors are as in 2A. Fig. 3. SV size distribution, sequence coverage, genes, and distribution of gene categories. (A) Size distribution of SVs (NA15510 and NA18505 combined). Arrow indicates the lower size cutoff for deletions. (B) Cumulative number of bp affected by SVs in relation to SV size (NA18505 only). (C) Solid line: cumulative number of RefSeq genes intersecting with SVs in relation to SV size (NA18505 only). Randomly shuffled SV locations within the local genomic context (±50kb window) exhibit an increase in gene overlap (dashed line). (D) Enrichment/depletion of Gene Ontology (GO; annotation level 3) biological processes for genes intersecting with SVs (NA15510 and NA18505 combined). Annotations / / 13 September 2007 / Page 5 / /science
6 represented by <10 genes are designated other and are gray. **denotes a significant enrichment in genes belonging to a category (P<1e-14; (12)); *significant depletion (P<0.001). Fig. 4. Validation of SVs. (A) A 170kb deletion detected with both array-cgh and PEM. (B) PCR products validating SVs as originally predicted from NA18505 (lane 2). Lanes 1-4 uses DNAs from NA15510, NA18505, NA11997 (HapMap CEU), and NA18614 (HapMap JCHI). Primer sequences can be found in table S6. (C) Fiber-FISH validation of heterozygous inversions in NA The inversion in the upper panel was independently validated in NA Alternating patterns of fluorescent labels from adjacent probes indicate genomic rearrangement. Fig. 5. Sequencing and analysis of SV breakpoint junctions. (A) PCR fragments spanning SVs were pooled and sequenced; breakpoints were determined from assembled contigs or 2 sequencing reads. (B) Representative sequenced SVs showing their relationship to previous SV/CNV assignments [earlier SV/CNV assignments often extend outside of the depicted regions (3, 4)]. From top to bottom: SVs resulting from NHEJ, L1 retrotransposition, HERVK (retrovirus) insertion, (non-allelic) homologous recombination, gap closure (blue: insertions, red: deletions, yellow: inversions). Note some SVs affect annotated genes. (C) Example breakpoint sequences (12). Upper case/green: unaltered sequence; lower case: 'SV indel'; solid arrows indicate microhomologies (indicative of NHEJ), duplication of target sequences (at retrotransposon/retrovirus insertion sites), and long stretches of sequence identity (12) (indicative of homologous recombination). Note that the fourth sequence (from top to bottom) shows an OR gene fusion in the main reading frame (breakpoints occured in the long stretch of sequence identity). / / 13 September 2007 / Page 6 / /science
7 Table 1. Validation of SVs identified by PEM. Array-CGH experiments were scored for indels in NA15510 not shared with NA An additional 88 SV breakpoint junctions were deduced from the Celera assembly (table S1). Est., estimation. Totals are underlined. Total SVs Intersection with SVs/CNVs in DGV SVs confirmed by Celera assembly Array- CGH Fiber-FISH (no. of validated inversions indicated) PCR spanning breakpoint junctions SV events with sequenced breakpoint junctions Female of presumably European ancestry (NA15510) SVs detected by PEM SV Indels Inversions SVs genomewide est Female of African ancestry (NA18505) SVs detected by PEM SV Indels Inversions SVs genomewide est
8
9
10

11
12
Structural variation. Marta Puig Institut de Biotecnologia i Biomedicina Universitat Autònoma de Barcelona
 Structural variation Marta Puig Institut de Biotecnologia i Biomedicina Universitat Autònoma de Barcelona Genetic variation How much genetic variation is there between individuals? What type of variants
Structural variation Marta Puig Institut de Biotecnologia i Biomedicina Universitat Autònoma de Barcelona Genetic variation How much genetic variation is there between individuals? What type of variants
Nature Biotechnology: doi: /nbt Supplementary Figure 1. Number and length distributions of the inferred fosmids.
 Supplementary Figure 1 Number and length distributions of the inferred fosmids. Fosmid were inferred by mapping each pool s sequence reads to hg19. We retained only those reads that mapped to within a
Supplementary Figure 1 Number and length distributions of the inferred fosmids. Fosmid were inferred by mapping each pool s sequence reads to hg19. We retained only those reads that mapped to within a
Mate-pair library data improves genome assembly
 De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
Figure S1. Unrearranged locus. Rearranged locus. Concordant read pairs. Region1. Region2. Cluster of discordant read pairs, bundle
 Figure S1 a Unrearranged locus Rearranged locus Concordant read pairs Region1 Concordant read pairs Cluster of discordant read pairs, bundle Region2 Concordant read pairs b Physical coverage 5 4 3 2 1
Figure S1 a Unrearranged locus Rearranged locus Concordant read pairs Region1 Concordant read pairs Cluster of discordant read pairs, bundle Region2 Concordant read pairs b Physical coverage 5 4 3 2 1
NUCLEOTIDE RESOLUTION STRUCTURAL VARIATION DETECTION USING NEXT- GENERATION WHOLE GENOME RESEQUENCING
 NUCLEOTIDE RESOLUTION STRUCTURAL VARIATION DETECTION USING NEXT- GENERATION WHOLE GENOME RESEQUENCING Ken Chen, Ph.D. kchen@genome.wustl.edu The Genome Center, Washington University in St. Louis The path
NUCLEOTIDE RESOLUTION STRUCTURAL VARIATION DETECTION USING NEXT- GENERATION WHOLE GENOME RESEQUENCING Ken Chen, Ph.D. kchen@genome.wustl.edu The Genome Center, Washington University in St. Louis The path
This is a closed book, closed note exam. No calculators, phones or any electronic device are allowed.
 MCB 104 MIDTERM #2 October 23, 2013 ***IMPORTANT REMINDERS*** Print your name and ID# on every page of the exam. You will lose 0.5 point/page if you forget to do this. Name KEY If you need more space than
MCB 104 MIDTERM #2 October 23, 2013 ***IMPORTANT REMINDERS*** Print your name and ID# on every page of the exam. You will lose 0.5 point/page if you forget to do this. Name KEY If you need more space than
Complementary Technologies for Precision Genetic Analysis
 Complementary NGS, CGH and Workflow Featured Publication Zhu, J. et al. Duplication of C7orf58, WNT16 and FAM3C in an obese female with a t(7;22)(q32.1;q11.2) chromosomal translocation and clinical features
Complementary NGS, CGH and Workflow Featured Publication Zhu, J. et al. Duplication of C7orf58, WNT16 and FAM3C in an obese female with a t(7;22)(q32.1;q11.2) chromosomal translocation and clinical features
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
GENE MAPPING. Genetica per Scienze Naturali a.a prof S. Presciuttini
 GENE MAPPING Questo documento è pubblicato sotto licenza Creative Commons Attribuzione Non commerciale Condividi allo stesso modo http://creativecommons.org/licenses/by-nc-sa/2.5/deed.it Genetic mapping
GENE MAPPING Questo documento è pubblicato sotto licenza Creative Commons Attribuzione Non commerciale Condividi allo stesso modo http://creativecommons.org/licenses/by-nc-sa/2.5/deed.it Genetic mapping
REVIEWS. Structural variation in the human genome
 REVIEWS Structural variation in the human genome Lars Feuk, Andrew R. Carson and Stephen W. Scherer Abstract The first wave of information from the analysis of the human genome revealed SNPs to be the
REVIEWS Structural variation in the human genome Lars Feuk, Andrew R. Carson and Stephen W. Scherer Abstract The first wave of information from the analysis of the human genome revealed SNPs to be the
BENG 183 Trey Ideker. Genome Assembly and Physical Mapping
 BENG 183 Trey Ideker Genome Assembly and Physical Mapping Reasons for sequencing Complete genome sequencing!!! Resequencing (Confirmatory) E.g., short regions containing single nucleotide polymorphisms
BENG 183 Trey Ideker Genome Assembly and Physical Mapping Reasons for sequencing Complete genome sequencing!!! Resequencing (Confirmatory) E.g., short regions containing single nucleotide polymorphisms
CS273B: Deep Learning in Genomics and Biomedicine. Recitation 1 30/9/2016
 CS273B: Deep Learning in Genomics and Biomedicine. Recitation 1 30/9/2016 Topics Genetic variation Population structure Linkage disequilibrium Natural disease variants Genome Wide Association Studies Gene
CS273B: Deep Learning in Genomics and Biomedicine. Recitation 1 30/9/2016 Topics Genetic variation Population structure Linkage disequilibrium Natural disease variants Genome Wide Association Studies Gene
Annotating Fosmid 14p24 of D. Virilis chromosome 4
 Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
FINDING THE PAIN GENE How do geneticists connect a specific gene with a specific phenotype?
 FINDING THE PAIN GENE How do geneticists connect a specific gene with a specific phenotype? 1 Linkage & Recombination HUH? What? Why? Who cares? How? Multiple choice question. Each colored line represents
FINDING THE PAIN GENE How do geneticists connect a specific gene with a specific phenotype? 1 Linkage & Recombination HUH? What? Why? Who cares? How? Multiple choice question. Each colored line represents
Lecture 2: Biology Basics Continued
 Lecture 2: Biology Basics Continued Central Dogma DNA: The Code of Life The structure and the four genomic letters code for all living organisms Adenine, Guanine, Thymine, and Cytosine which pair A-T and
Lecture 2: Biology Basics Continued Central Dogma DNA: The Code of Life The structure and the four genomic letters code for all living organisms Adenine, Guanine, Thymine, and Cytosine which pair A-T and
Phasing of 2-SNP Genotypes based on Non-Random Mating Model
 Phasing of 2-SNP Genotypes based on Non-Random Mating Model Dumitru Brinza and Alexander Zelikovsky Department of Computer Science, Georgia State University, Atlanta, GA 30303 {dima,alexz}@cs.gsu.edu Abstract.
Phasing of 2-SNP Genotypes based on Non-Random Mating Model Dumitru Brinza and Alexander Zelikovsky Department of Computer Science, Georgia State University, Atlanta, GA 30303 {dima,alexz}@cs.gsu.edu Abstract.
Chapter 6 - Molecular Genetic Techniques
 Chapter 6 - Molecular Genetic Techniques Two objects of molecular & genetic technologies For analysis For generation Molecular genetic technologies! For analysis DNA gel electrophoresis Southern blotting
Chapter 6 - Molecular Genetic Techniques Two objects of molecular & genetic technologies For analysis For generation Molecular genetic technologies! For analysis DNA gel electrophoresis Southern blotting
Initial sequence of the chimpanzee genome and comparison with the human genome
 Vol 437 1 September 2005 doi:10.1038/nature04072 Initial sequence of the chimpanzee genome and comparison with the human genome The Chimpanzee Sequencing and Analysis Consortium* Here we present a draft
Vol 437 1 September 2005 doi:10.1038/nature04072 Initial sequence of the chimpanzee genome and comparison with the human genome The Chimpanzee Sequencing and Analysis Consortium* Here we present a draft
Introduction to RNA-Seq. David Wood Winter School in Mathematics and Computational Biology July 1, 2013
 Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Computational methods for discovering structural variation with next-generation sequencing
 Computational methods for discovering structural variation with next-generation sequencing Paul Medvedev 1, Monica Stanciu 1 & Michael Brudno 1,2 In the last several years, a number of studies have described
Computational methods for discovering structural variation with next-generation sequencing Paul Medvedev 1, Monica Stanciu 1 & Michael Brudno 1,2 In the last several years, a number of studies have described
Amapofhumangenomevariationfrom population-scale sequencing
 doi:.38/nature9534 Amapofhumangenomevariationfrom population-scale sequencing The Genomes Project Consortium* The Genomes Project aims to provide a deep characterization of human genome sequence variation
doi:.38/nature9534 Amapofhumangenomevariationfrom population-scale sequencing The Genomes Project Consortium* The Genomes Project aims to provide a deep characterization of human genome sequence variation
An introduction to genetics and molecular biology
 An introduction to genetics and molecular biology Cavan Reilly September 5, 2017 Table of contents Introduction to biology Some molecular biology Gene expression Mendelian genetics Some more molecular
An introduction to genetics and molecular biology Cavan Reilly September 5, 2017 Table of contents Introduction to biology Some molecular biology Gene expression Mendelian genetics Some more molecular
Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4
 WHITE PAPER Oncomine Comprehensive Assay Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4 Contents Scope and purpose of document...2 Content...2 How Torrent
WHITE PAPER Oncomine Comprehensive Assay Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4 Contents Scope and purpose of document...2 Content...2 How Torrent
Sept 2. Structure and Organization of Genomes. Today: Genetic and Physical Mapping. Sept 9. Forward and Reverse Genetics. Genetic and Physical Mapping
 Sept 2. Structure and Organization of Genomes Today: Genetic and Physical Mapping Assignments: Gibson & Muse, pp.4-10 Brown, pp. 126-160 Olson et al., Science 245: 1434 New homework:due, before class,
Sept 2. Structure and Organization of Genomes Today: Genetic and Physical Mapping Assignments: Gibson & Muse, pp.4-10 Brown, pp. 126-160 Olson et al., Science 245: 1434 New homework:due, before class,
Human SNP haplotypes. Statistics 246, Spring 2002 Week 15, Lecture 1
 Human SNP haplotypes Statistics 246, Spring 2002 Week 15, Lecture 1 Human single nucleotide polymorphisms The majority of human sequence variation is due to substitutions that have occurred once in the
Human SNP haplotypes Statistics 246, Spring 2002 Week 15, Lecture 1 Human single nucleotide polymorphisms The majority of human sequence variation is due to substitutions that have occurred once in the
Sequence assembly. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
Finishing Fosmid DMAC-27a of the Drosophila mojavensis third chromosome
 Finishing Fosmid DMAC-27a of the Drosophila mojavensis third chromosome Ruth Howe Bio 434W 27 February 2010 Abstract The fourth or dot chromosome of Drosophila species is composed primarily of highly condensed,
Finishing Fosmid DMAC-27a of the Drosophila mojavensis third chromosome Ruth Howe Bio 434W 27 February 2010 Abstract The fourth or dot chromosome of Drosophila species is composed primarily of highly condensed,
Assignment 9: Genetic Variation
 Assignment 9: Genetic Variation Due Date: Friday, March 30 th, 2018, 10 am In this assignment, you will profile genome variation information and attempt to answer biologically relevant questions. The variant
Assignment 9: Genetic Variation Due Date: Friday, March 30 th, 2018, 10 am In this assignment, you will profile genome variation information and attempt to answer biologically relevant questions. The variant
Overview of Human Genetics
 Overview of Human Genetics 1 Structure and function of nucleic acids. 2 Structure and composition of the human genome. 3 Mendelian genetics. Lander et al. (Nature, 2001) MAT 394 (ASU) Human Genetics Spring
Overview of Human Genetics 1 Structure and function of nucleic acids. 2 Structure and composition of the human genome. 3 Mendelian genetics. Lander et al. (Nature, 2001) MAT 394 (ASU) Human Genetics Spring
Genomes summary. Bacterial genome sizes
 Genomes summary 1. >930 bacterial genomes sequenced. 2. Circular. Genes densely packed. 3. 2-10 Mbases, 470-7,000 genes 4. Genomes of >200 eukaryotes (45 higher ) sequenced. 5. Linear chromosomes 6. On
Genomes summary 1. >930 bacterial genomes sequenced. 2. Circular. Genes densely packed. 3. 2-10 Mbases, 470-7,000 genes 4. Genomes of >200 eukaryotes (45 higher ) sequenced. 5. Linear chromosomes 6. On
Nature Biotechnology: doi: /nbt.3943
 Supplementary Figure 1. Distribution of sequence depth across the bacterial artificial chromosomes (BACs). The x-axis denotes the sequencing depth (X) of each BAC and y-axis denotes the number of BACs
Supplementary Figure 1. Distribution of sequence depth across the bacterial artificial chromosomes (BACs). The x-axis denotes the sequencing depth (X) of each BAC and y-axis denotes the number of BACs
Single Nucleotide Variant Analysis. H3ABioNet May 14, 2014
 Single Nucleotide Variant Analysis H3ABioNet May 14, 2014 Outline What are SNPs and SNVs? How do we identify them? How do we call them? SAMTools GATK VCF File Format Let s call variants! Single Nucleotide
Single Nucleotide Variant Analysis H3ABioNet May 14, 2014 Outline What are SNPs and SNVs? How do we identify them? How do we call them? SAMTools GATK VCF File Format Let s call variants! Single Nucleotide
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
Gene Expression Technology
 Gene Expression Technology Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Gene expression Gene expression is the process by which information from a gene
Gene Expression Technology Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Gene expression Gene expression is the process by which information from a gene
What is genetic variation?
 enetic Variation Applied Computational enomics, Lecture 05 https://github.com/quinlan-lab/applied-computational-genomics Aaron Quinlan Departments of Human enetics and Biomedical Informatics USTAR Center
enetic Variation Applied Computational enomics, Lecture 05 https://github.com/quinlan-lab/applied-computational-genomics Aaron Quinlan Departments of Human enetics and Biomedical Informatics USTAR Center
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Supplementary Material
 Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Methods and strategies for analyzing copy number variation using DNA microarrays
 Methods and strategies for analyzing copy number variation using DNA microarrays Nigel P Carter The association of DNA copy-number variation (CNV) with specific gene function and human disease has been
Methods and strategies for analyzing copy number variation using DNA microarrays Nigel P Carter The association of DNA copy-number variation (CNV) with specific gene function and human disease has been
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
Development and application of CGHPRO, a novel software package for retrieving, handling and analysing array CGH data
 Aus dem Max Planck Institut für molekulare Genetik DISSERTATION Development and application of CGHPRO, a novel software package for retrieving, handling and analysing array CGH data Zur Erlangung des akademischen
Aus dem Max Planck Institut für molekulare Genetik DISSERTATION Development and application of CGHPRO, a novel software package for retrieving, handling and analysing array CGH data Zur Erlangung des akademischen
Genome-wide association studies (GWAS) Part 1
 Part 1") Genome-wide association studies (GWAS) Part 1 Matti Pirinen FIMM, University of Helsinki 03.12.2013, Kumpula Campus FIMM - Institiute for Molecular Medicine Finland www.fimm.fi Published Genome-Wide Associations
Genome-wide association studies (GWAS) Part 1 Matti Pirinen FIMM, University of Helsinki 03.12.2013, Kumpula Campus FIMM - Institiute for Molecular Medicine Finland www.fimm.fi Published Genome-Wide Associations
DNA Collection. Data Quality Control. Whole Genome Amplification. Whole Genome Amplification. Measure DNA concentrations. Pros
 DNA Collection Data Quality Control Suzanne M. Leal Baylor College of Medicine sleal@bcm.edu Copyrighted S.M. Leal 2016 Blood samples For unlimited supply of DNA Transformed cell lines Buccal Swabs Small
DNA Collection Data Quality Control Suzanne M. Leal Baylor College of Medicine sleal@bcm.edu Copyrighted S.M. Leal 2016 Blood samples For unlimited supply of DNA Transformed cell lines Buccal Swabs Small
Finishing Drosophila Ananassae Fosmid 2728G16
 Finishing Drosophila Ananassae Fosmid 2728G16 Kyle Jung March 8, 2013 Bio434W Professor Elgin Page 1 Abstract For my finishing project, I chose to finish fosmid 2728G16. This fosmid carries a segment of
Finishing Drosophila Ananassae Fosmid 2728G16 Kyle Jung March 8, 2013 Bio434W Professor Elgin Page 1 Abstract For my finishing project, I chose to finish fosmid 2728G16. This fosmid carries a segment of
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature09937 a Name Position Primersets 1a 1b 2 3 4 b2 Phenotype Genotype b Primerset 1a D T C R I E 10000 8000 6000 5000 4000 3000 2500 2000 1500 1000 800 Donor (D)
SUPPLEMENTARY INFORMATION doi:10.1038/nature09937 a Name Position Primersets 1a 1b 2 3 4 b2 Phenotype Genotype b Primerset 1a D T C R I E 10000 8000 6000 5000 4000 3000 2500 2000 1500 1000 800 Donor (D)
Axiom mydesign Custom Array design guide for human genotyping applications
 TECHNICAL NOTE Axiom mydesign Custom Genotyping Arrays Axiom mydesign Custom Array design guide for human genotyping applications Overview In the past, custom genotyping arrays were expensive, required
TECHNICAL NOTE Axiom mydesign Custom Genotyping Arrays Axiom mydesign Custom Array design guide for human genotyping applications Overview In the past, custom genotyping arrays were expensive, required
Genome-Wide Association Studies (GWAS): Computational Them
: Computational Them") Genome-Wide Association Studies (GWAS): Computational Themes and Caveats October 14, 2014 Many issues in Genomewide Association Studies We show that even for the simplest analysis, there is little consensus
Genome-Wide Association Studies (GWAS): Computational Themes and Caveats October 14, 2014 Many issues in Genomewide Association Studies We show that even for the simplest analysis, there is little consensus
Chapter 4 Gene Linkage and Genetic Mapping
 Chapter 4 Gene Linkage and Genetic Mapping 1 Important Definitions Locus = physical location of a gene on a chromosome Homologous pairs of chromosomes often contain alternative forms of a given gene =
Chapter 4 Gene Linkage and Genetic Mapping 1 Important Definitions Locus = physical location of a gene on a chromosome Homologous pairs of chromosomes often contain alternative forms of a given gene =
Enhancers mutations that make the original mutant phenotype more extreme. Suppressors mutations that make the original mutant phenotype less extreme
 Interactomics and Proteomics 1. Interactomics The field of interactomics is concerned with interactions between genes or proteins. They can be genetic interactions, in which two genes are involved in the
Interactomics and Proteomics 1. Interactomics The field of interactomics is concerned with interactions between genes or proteins. They can be genetic interactions, in which two genes are involved in the
7 Gene Isolation and Analysis of Multiple
 Genetic Techniques for Biological Research Corinne A. Michels Copyright q 2002 John Wiley & Sons, Ltd ISBNs: 0-471-89921-6 (Hardback); 0-470-84662-3 (Electronic) 7 Gene Isolation and Analysis of Multiple
Genetic Techniques for Biological Research Corinne A. Michels Copyright q 2002 John Wiley & Sons, Ltd ISBNs: 0-471-89921-6 (Hardback); 0-470-84662-3 (Electronic) 7 Gene Isolation and Analysis of Multiple
Sequence Variations. Baxevanis and Ouellette, Chapter 7 - Sequence Polymorphisms. NCBI SNP Primer:
 Sequence Variations Baxevanis and Ouellette, Chapter 7 - Sequence Polymorphisms NCBI SNP Primer: http://www.ncbi.nlm.nih.gov/about/primer/snps.html Overview Mutation and Alleles Linkage Genetic variation
Sequence Variations Baxevanis and Ouellette, Chapter 7 - Sequence Polymorphisms NCBI SNP Primer: http://www.ncbi.nlm.nih.gov/about/primer/snps.html Overview Mutation and Alleles Linkage Genetic variation
Using mutants to clone genes
 Using mutants to clone genes Objectives 1. What is positional cloning? 2. What is insertional tagging? 3. How can one confirm that the gene cloned is the same one that is mutated to give the phenotype
Using mutants to clone genes Objectives 1. What is positional cloning? 2. What is insertional tagging? 3. How can one confirm that the gene cloned is the same one that is mutated to give the phenotype
AGRO/ANSC/BIO/GENE/HORT 305 Fall, 2016 Overview of Genetics Lecture outline (Chpt 1, Genetics by Brooker) #1
 #1") AGRO/ANSC/BIO/GENE/HORT 305 Fall, 2016 Overview of Genetics Lecture outline (Chpt 1, Genetics by Brooker) #1 - Genetics: Progress from Mendel to DNA: Gregor Mendel, in the mid 19 th century provided the
AGRO/ANSC/BIO/GENE/HORT 305 Fall, 2016 Overview of Genetics Lecture outline (Chpt 1, Genetics by Brooker) #1 - Genetics: Progress from Mendel to DNA: Gregor Mendel, in the mid 19 th century provided the
Conifer Translational Genomics Network Coordinated Agricultural Project
 Conifer Translational Genomics Network Coordinated Agricultural Project Genomics in Tree Breeding and Forest Ecosystem Management ----- Module 2 Genes, Genomes, and Mendel Nicholas Wheeler & David Harry
Conifer Translational Genomics Network Coordinated Agricultural Project Genomics in Tree Breeding and Forest Ecosystem Management ----- Module 2 Genes, Genomes, and Mendel Nicholas Wheeler & David Harry
Mutagenesis for Studying Gene Function Spring, 2007 Guangyi Wang, Ph.D. POST103B
 Mutagenesis for Studying Gene Function Spring, 2007 Guangyi Wang, Ph.D. POST103B guangyi@hawaii.edu http://www.soest.hawaii.edu/marinefungi/ocn403webpage.htm Overview of Last Lecture DNA microarray hybridization
Mutagenesis for Studying Gene Function Spring, 2007 Guangyi Wang, Ph.D. POST103B guangyi@hawaii.edu http://www.soest.hawaii.edu/marinefungi/ocn403webpage.htm Overview of Last Lecture DNA microarray hybridization
GREG GIBSON SPENCER V. MUSE
 A Primer of Genome Science ience THIRD EDITION TAGCACCTAGAATCATGGAGAGATAATTCGGTGAGAATTAAATGGAGAGTTGCATAGAGAACTGCGAACTG GREG GIBSON SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc.
A Primer of Genome Science ience THIRD EDITION TAGCACCTAGAATCATGGAGAGATAATTCGGTGAGAATTAAATGGAGAGTTGCATAGAGAACTGCGAACTG GREG GIBSON SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc.
High-Resolution Oligonucleotide- Based acgh Analysis of Single Cells in Under 24 Hours
 High-Resolution Oligonucleotide- Based acgh Analysis of Single Cells in Under 24 Hours Application Note Authors Paula Costa and Anniek De Witte Agilent Technologies, Inc. Santa Clara, CA USA Abstract As
High-Resolution Oligonucleotide- Based acgh Analysis of Single Cells in Under 24 Hours Application Note Authors Paula Costa and Anniek De Witte Agilent Technologies, Inc. Santa Clara, CA USA Abstract As
Theoretische Biologie
 Theoretische Biologie Prof. Computational EvoDevo, University of Leipzig SS 2017 Two Gene Concepts in Comparison Gerstein-Snyder gene definition Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel
Theoretische Biologie Prof. Computational EvoDevo, University of Leipzig SS 2017 Two Gene Concepts in Comparison Gerstein-Snyder gene definition Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel
Mapping and Mapping Populations
 Mapping and Mapping Populations Types of mapping populations F 2 o Two F 1 individuals are intermated Backcross o Cross of a recurrent parent to a F 1 Recombinant Inbred Lines (RILs; F 2 -derived lines)
Mapping and Mapping Populations Types of mapping populations F 2 o Two F 1 individuals are intermated Backcross o Cross of a recurrent parent to a F 1 Recombinant Inbred Lines (RILs; F 2 -derived lines)
Midterm 1 Results. Midterm 1 Akey/ Fields Median Number of Students. Exam Score
 Midterm 1 Results 10 Midterm 1 Akey/ Fields Median - 69 8 Number of Students 6 4 2 0 21 26 31 36 41 46 51 56 61 66 71 76 81 86 91 96 101 Exam Score Quick review of where we left off Parental type: the
Midterm 1 Results 10 Midterm 1 Akey/ Fields Median - 69 8 Number of Students 6 4 2 0 21 26 31 36 41 46 51 56 61 66 71 76 81 86 91 96 101 Exam Score Quick review of where we left off Parental type: the
Mutations during meiosis and germ line division lead to genetic variation between individuals
 Mutations during meiosis and germ line division lead to genetic variation between individuals Types of mutations: point mutations indels (insertion/deletion) copy number variation structural rearrangements
Mutations during meiosis and germ line division lead to genetic variation between individuals Types of mutations: point mutations indels (insertion/deletion) copy number variation structural rearrangements
T and B cell gene rearrangement October 17, Ram Savan
 T and B cell gene rearrangement October 17, 2016 Ram Savan savanram@uw.edu 441 Lecture #9 Slide 1 of 28 Three lectures on antigen receptors Part 1 (Last Friday): Structural features of the BCR and TCR
T and B cell gene rearrangement October 17, 2016 Ram Savan savanram@uw.edu 441 Lecture #9 Slide 1 of 28 Three lectures on antigen receptors Part 1 (Last Friday): Structural features of the BCR and TCR
1. A brief overview of sequencing biochemistry
 Supplementary reading materials on Genome sequencing (optional) The materials are from Mark Blaxter s lecture notes on Sequencing strategies and Primary Analysis 1. A brief overview of sequencing biochemistry
Supplementary reading materials on Genome sequencing (optional) The materials are from Mark Blaxter s lecture notes on Sequencing strategies and Primary Analysis 1. A brief overview of sequencing biochemistry
Measurement of Molecular Genetic Variation. Forces Creating Genetic Variation. Mutation: Nucleotide Substitutions
 Measurement of Molecular Genetic Variation Genetic Variation Is The Necessary Prerequisite For All Evolution And For Studying All The Major Problem Areas In Molecular Evolution. How We Score And Measure
Measurement of Molecular Genetic Variation Genetic Variation Is The Necessary Prerequisite For All Evolution And For Studying All The Major Problem Areas In Molecular Evolution. How We Score And Measure
Supplementary Figures
 Supplementary Figures 1 Supplementary Figure 1. Analyses of present-day population differentiation. (A, B) Enrichment of strongly differentiated genic alleles for all present-day population comparisons
Supplementary Figures 1 Supplementary Figure 1. Analyses of present-day population differentiation. (A, B) Enrichment of strongly differentiated genic alleles for all present-day population comparisons
Welcome to the NGS webinar series
 Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
Comparative Genomic Hybridization
 Comparative Genomic Hybridization Srikesh G. Arunajadai Division of Biostatistics University of California Berkeley PH 296 Presentation Fall 2002 December 9 th 2002 OUTLINE CGH Introduction Methodology,
Comparative Genomic Hybridization Srikesh G. Arunajadai Division of Biostatistics University of California Berkeley PH 296 Presentation Fall 2002 December 9 th 2002 OUTLINE CGH Introduction Methodology,
Human genetic variation
 Human genetic variation CHEW Fook Tim Human Genetic Variation Variants contribute to rare and common diseases Variants can be used to trace human origins Human Genetic Variation What types of variants
Human genetic variation CHEW Fook Tim Human Genetic Variation Variants contribute to rare and common diseases Variants can be used to trace human origins Human Genetic Variation What types of variants
The human noncoding genome defined by genetic diversity
 SUPPLEMENTARY INFORMATION Letters https://doi.org/10.1038/s41588-018-0062-7 In the format provided by the authors and unedited. The human noncoding genome defined by genetic diversity Julia di Iulio 1,5,
SUPPLEMENTARY INFORMATION Letters https://doi.org/10.1038/s41588-018-0062-7 In the format provided by the authors and unedited. The human noncoding genome defined by genetic diversity Julia di Iulio 1,5,
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Genome Sequence Assembly
 Genome Sequence Assembly Learning Goals: Introduce the field of bioinformatics Familiarize the student with performing sequence alignments Understand the assembly process in genome sequencing Introduction:
Genome Sequence Assembly Learning Goals: Introduce the field of bioinformatics Familiarize the student with performing sequence alignments Understand the assembly process in genome sequencing Introduction:
Cancer Genetics Solutions
 Cancer Genetics Solutions Cancer Genetics Solutions Pushing the Boundaries in Cancer Genetics Cancer is a formidable foe that presents significant challenges. The complexity of this disease can be daunting
Cancer Genetics Solutions Cancer Genetics Solutions Pushing the Boundaries in Cancer Genetics Cancer is a formidable foe that presents significant challenges. The complexity of this disease can be daunting
Personal Genomics Platform White Paper Last Updated November 15, Executive Summary
 Executive Summary Helix is a personal genomics platform company with a simple but powerful mission: to empower every person to improve their life through DNA. Our platform includes saliva sample collection,
Executive Summary Helix is a personal genomics platform company with a simple but powerful mission: to empower every person to improve their life through DNA. Our platform includes saliva sample collection,
Genetics and Genomics in Medicine Chapter 3. Questions & Answers
 Genetics and Genomics in Medicine Chapter 3 Multiple Choice Questions Questions & Answers Question 3.1 Which of the following statements, if any, is false? a) Amplifying DNA means making many identical
Genetics and Genomics in Medicine Chapter 3 Multiple Choice Questions Questions & Answers Question 3.1 Which of the following statements, if any, is false? a) Amplifying DNA means making many identical
3. human genomics clone genes associated with genetic disorders. 4. many projects generate ordered clones that cover genome
 Lectures 30 and 31 Genome analysis I. Genome analysis A. two general areas 1. structural 2. functional B. genome projects a status report 1. 1 st sequenced: several viral genomes 2. mitochondria and chloroplasts
Lectures 30 and 31 Genome analysis I. Genome analysis A. two general areas 1. structural 2. functional B. genome projects a status report 1. 1 st sequenced: several viral genomes 2. mitochondria and chloroplasts
SNPs - GWAS - eqtls. Sebastian Schmeier
 SNPs - GWAS - eqtls s.schmeier@gmail.com http://sschmeier.github.io/bioinf-workshop/ 17.08.2015 Overview Single nucleotide polymorphism (refresh) SNPs effect on genes (refresh) Genome-wide association
SNPs - GWAS - eqtls s.schmeier@gmail.com http://sschmeier.github.io/bioinf-workshop/ 17.08.2015 Overview Single nucleotide polymorphism (refresh) SNPs effect on genes (refresh) Genome-wide association
Release Notes for Genomes Processed Using Complete Genomics Software
 Release Notes for Genomes Processed Using Complete Genomics Software Version 1.11.0 Related Documents... 1 Changes to Version 1.11.0... 2 Changes to Version 1.10.0... 6 Changes to Version 1.9.0... 10 Changes
Release Notes for Genomes Processed Using Complete Genomics Software Version 1.11.0 Related Documents... 1 Changes to Version 1.11.0... 2 Changes to Version 1.10.0... 6 Changes to Version 1.9.0... 10 Changes
Jenny Gu, PhD Strategic Business Development Manager, PacBio
 IDT and PacBio joint presentation Characterizing Alzheimer s Disease candidate genes and transcripts with targeted, long-read, single-molecule sequencing Jenny Gu, PhD Strategic Business Development Manager,
IDT and PacBio joint presentation Characterizing Alzheimer s Disease candidate genes and transcripts with targeted, long-read, single-molecule sequencing Jenny Gu, PhD Strategic Business Development Manager,
SUPPLEMENTAL MATERIALS
 SUPPLEMENL MERILS Eh-seq: RISPR epitope tagging hip-seq of DN-binding proteins Daniel Savic, E. hristopher Partridge, Kimberly M. Newberry, Sophia. Smith, Sarah K. Meadows, rian S. Roberts, Mark Mackiewicz,
SUPPLEMENL MERILS Eh-seq: RISPR epitope tagging hip-seq of DN-binding proteins Daniel Savic, E. hristopher Partridge, Kimberly M. Newberry, Sophia. Smith, Sarah K. Meadows, rian S. Roberts, Mark Mackiewicz,
Genetic Variation and Genome- Wide Association Studies. Keyan Salari, MD/PhD Candidate Department of Genetics
 Genetic Variation and Genome- Wide Association Studies Keyan Salari, MD/PhD Candidate Department of Genetics How many of you did the readings before class? A. Yes, of course! B. Started, but didn t get
Genetic Variation and Genome- Wide Association Studies Keyan Salari, MD/PhD Candidate Department of Genetics How many of you did the readings before class? A. Yes, of course! B. Started, but didn t get
Next Generation Sequencing. Target Enrichment
 Next Generation Sequencing Target Enrichment Next Generation Sequencing Your Partner in Every Step from Sample to Data NGS: Revolutionizing Genetic Analysis with Single-Molecule Resolution Next generation
Next Generation Sequencing Target Enrichment Next Generation Sequencing Your Partner in Every Step from Sample to Data NGS: Revolutionizing Genetic Analysis with Single-Molecule Resolution Next generation
Molecular Biology: DNA sequencing
 Molecular Biology: DNA sequencing Author: Prof Marinda Oosthuizen Licensed under a Creative Commons Attribution license. SEQUENCING OF LARGE TEMPLATES As we have seen, we can obtain up to 800 nucleotides
Molecular Biology: DNA sequencing Author: Prof Marinda Oosthuizen Licensed under a Creative Commons Attribution license. SEQUENCING OF LARGE TEMPLATES As we have seen, we can obtain up to 800 nucleotides
A Modified Digestion-Circularization PCR (DC-PCR) Approach to Detect Hypermutation- Associated DNA Double-Strand Breaks
 Approach to Detect Hypermutation- Associated DNA Double-Strand Breaks") A Modified Digestion-Circularization PCR (DC-PCR) Approach to Detect Hypermutation- Associated DNA Double-Strand Breaks SARAH K. DICKERSON AND F. NINA PAPAVASILIOU Laboratory of Lymphocyte Biology, The
A Modified Digestion-Circularization PCR (DC-PCR) Approach to Detect Hypermutation- Associated DNA Double-Strand Breaks SARAH K. DICKERSON AND F. NINA PAPAVASILIOU Laboratory of Lymphocyte Biology, The
Target Enrichment Strategies for Next Generation Sequencing
 Target Enrichment Strategies for Next Generation Sequencing Anuj Gupta, PhD Agilent Technologies, New Delhi Genotypic Conference, Sept 2014 NGS Timeline Information burst Nearly 30,000 human genomes sequenced
Target Enrichment Strategies for Next Generation Sequencing Anuj Gupta, PhD Agilent Technologies, New Delhi Genotypic Conference, Sept 2014 NGS Timeline Information burst Nearly 30,000 human genomes sequenced
Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John DiCarlo Yexun Wang
 Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Suggest a technique that could be used to provide molecular evidence that all English Elm trees form a clone. ... [1]
![Suggest a technique that could be used to provide molecular evidence that all English Elm trees form a clone. ... [1]](/thumbs/77/74807071.jpg "Suggest a technique that could be used to provide molecular evidence that all English Elm trees form a clone. ... [1]") 1 Molecular evidence E Ulmus procera, form a genetically isolated clone. English Elms developed from a variety of elm brought to Britain from Rome in the first century A.D. Although English Elm trees make
1 Molecular evidence E Ulmus procera, form a genetically isolated clone. English Elms developed from a variety of elm brought to Britain from Rome in the first century A.D. Although English Elm trees make
Chapter 15 Gene Technologies and Human Applications
 Chapter Outline Chapter 15 Gene Technologies and Human Applications Section 1: The Human Genome KEY IDEAS > Why is the Human Genome Project so important? > How do genomics and gene technologies affect
Chapter Outline Chapter 15 Gene Technologies and Human Applications Section 1: The Human Genome KEY IDEAS > Why is the Human Genome Project so important? > How do genomics and gene technologies affect
User Manual. Catalog No.: DWSK-V101 (10 rxns), DWSK-V102 (25 rxns) For Research Use Only
, DWSK-V102 (25 rxns) For Research Use Only") DNA Walking SpeedUp TM Kit SpeedUp Sequencing SpeedUp BAC Clone Sequencing SpeedUp Genome Walking SpeedUp Transgene Location Detection SpeedUp Deletion/ Insertion/ Isoform Detection User Manual Version
DNA Walking SpeedUp TM Kit SpeedUp Sequencing SpeedUp BAC Clone Sequencing SpeedUp Genome Walking SpeedUp Transgene Location Detection SpeedUp Deletion/ Insertion/ Isoform Detection User Manual Version
Supplementary Figures Montero et al._supplementary Figure 1
 Montero et al_suppl. Info 1 Supplementary Figures Montero et al._supplementary Figure 1 Montero et al_suppl. Info 2 Supplementary Figure 1. Transcripts arising from the structurally conserved subtelomeres
Montero et al_suppl. Info 1 Supplementary Figures Montero et al._supplementary Figure 1 Montero et al_suppl. Info 2 Supplementary Figure 1. Transcripts arising from the structurally conserved subtelomeres
Petar Pajic 1 *, Yen Lung Lin 1 *, Duo Xu 1, Omer Gokcumen 1 Department of Biological Sciences, University at Buffalo, Buffalo, NY.
 The psoriasis associated deletion of late cornified envelope genes LCE3B and LCE3C has been maintained under balancing selection since Human Denisovan divergence Petar Pajic 1 *, Yen Lung Lin 1 *, Duo
The psoriasis associated deletion of late cornified envelope genes LCE3B and LCE3C has been maintained under balancing selection since Human Denisovan divergence Petar Pajic 1 *, Yen Lung Lin 1 *, Duo
MoGUL: Detecting Common Insertions and Deletions in a Population
 MoGUL: Detecting Common Insertions and Deletions in a Population Seunghak Lee 1,2, Eric Xing 2, and Michael Brudno 1,3, 1 Department of Computer Science, University of Toronto, Canada 2 School of Computer
MoGUL: Detecting Common Insertions and Deletions in a Population Seunghak Lee 1,2, Eric Xing 2, and Michael Brudno 1,3, 1 Department of Computer Science, University of Toronto, Canada 2 School of Computer
Genome Sequencing-- Strategies
 Genome Sequencing-- Strategies Bio 4342 Spring 04 What is a genome? A genome can be defined as the entire DNA content of each nucleated cell in an organism Each organism has one or more chromosomes that
Genome Sequencing-- Strategies Bio 4342 Spring 04 What is a genome? A genome can be defined as the entire DNA content of each nucleated cell in an organism Each organism has one or more chromosomes that
5/18/2017. Genotypic, phenotypic or allelic frequencies each sum to 1. Changes in allele frequencies determine gene pool composition over generations
 Topics How to track evolution allele frequencies Hardy Weinberg principle applications Requirements for genetic equilibrium Types of natural selection Population genetic polymorphism in populations, pp.
Topics How to track evolution allele frequencies Hardy Weinberg principle applications Requirements for genetic equilibrium Types of natural selection Population genetic polymorphism in populations, pp.
De novo Genome Assembly
 De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
Structural Variation of the Human Genome
 Annu. Rev. Genomics Hum. Genet. 2006. 7:407 42 First published online as a Review in Advance on June 16, 2006 The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org This
Annu. Rev. Genomics Hum. Genet. 2006. 7:407 42 First published online as a Review in Advance on June 16, 2006 The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org This
Reading Lecture 8: Lecture 9: Lecture 8. DNA Libraries. Definition Types Construction
 Lecture 8 Reading Lecture 8: 96-110 Lecture 9: 111-120 DNA Libraries Definition Types Construction 142 DNA Libraries A DNA library is a collection of clones of genomic fragments or cdnas from a certain
Lecture 8 Reading Lecture 8: 96-110 Lecture 9: 111-120 DNA Libraries Definition Types Construction 142 DNA Libraries A DNA library is a collection of clones of genomic fragments or cdnas from a certain
Introduction. Thomas Hunt Morgan. Chromosomes and Inheritance. Drosophila melanogaster
 Chromosomes and Inheritance 1 4 Fig. 12-10, p. 244 Introduction It was not until 1900 that biology finally caught up with Gregor Mendel. Independently, Karl Correns, Erich von Tschermak, and Hugo de Vries
Chromosomes and Inheritance 1 4 Fig. 12-10, p. 244 Introduction It was not until 1900 that biology finally caught up with Gregor Mendel. Independently, Karl Correns, Erich von Tschermak, and Hugo de Vries
Construction of plant complementation vector and generation of transgenic plants
 MATERIAL S AND METHODS Plant materials and growth conditions Arabidopsis ecotype Columbia (Col0) was used for this study. SALK_072009, SALK_076309, and SALK_027645 were obtained from the Arabidopsis Biological
MATERIAL S AND METHODS Plant materials and growth conditions Arabidopsis ecotype Columbia (Col0) was used for this study. SALK_072009, SALK_076309, and SALK_027645 were obtained from the Arabidopsis Biological
Chimp Chunk 3-14 Annotation by Matthew Kwong, Ruth Howe, and Hao Yang
 Chimp Chunk 3-14 Annotation by Matthew Kwong, Ruth Howe, and Hao Yang Ruth Howe Bio 434W April 1, 2010 INTRODUCTION De novo annotation is the process by which a finished genomic sequence is searched for
Chimp Chunk 3-14 Annotation by Matthew Kwong, Ruth Howe, and Hao Yang Ruth Howe Bio 434W April 1, 2010 INTRODUCTION De novo annotation is the process by which a finished genomic sequence is searched for
Supplementary Figures
 Supplementary Figures Supplementary Fig. S1 Diagram of Pst genome sequencing and assembly using a fosmid to fosmid strategy. Fosmid pooling and sequencing: Fosmid librarywas constructed according to Kim
Supplementary Figures Supplementary Fig. S1 Diagram of Pst genome sequencing and assembly using a fosmid to fosmid strategy. Fosmid pooling and sequencing: Fosmid librarywas constructed according to Kim