BLAST. compared with database sequences Sequences with many matches to high- scoring words are used for final alignments
|
|
|
- Barbra Johnston
- 6 years ago
- Views:
Transcription
1 BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database sequences Sequences with many matches to high- scoring words are used for final alignments Protein based searches are always more powerful than nucleotide-base of coding g DNA in determining similarity and inferring homology

2 BLAST (Basic Local Alignment Search Tool) P=7+ Q=5 + G=6 In addition to the exact word, BLAST considers related words based on BLOSUM62: the neighborhood. Once a word is aligned, gapped and un-gapped extensions are initiated, tallying the cumulative score When the score drops more than X, the extension is terminated The extension is trimmed back to the maximum HSP= High scoring segment pair Produces local alignments X= significance decay S= min. score to return a BLAST hit T= neighborhood score threshold

3 BLAST home page
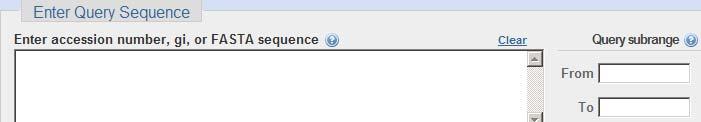
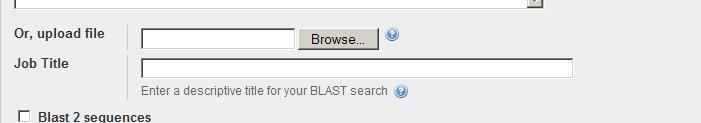
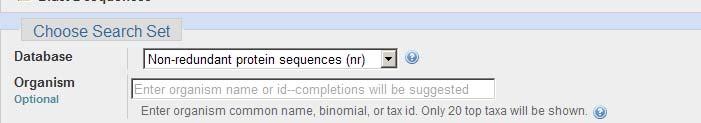
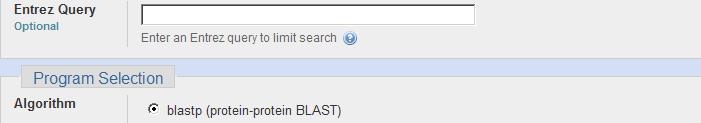
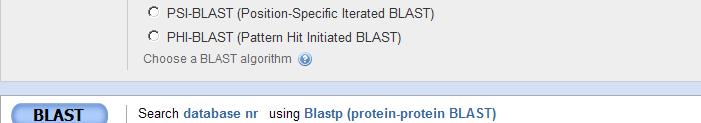

4 BLASTP


5 BLAST databases Peptide Sequence Databases nr: non-redundant GenBank CDS translations+pdb+swissprot+pir+prf RefSeq_protein: reference proteins Swissprot: SWISS-PROT protein sequence database pdb: Sequences derived from the 3-dimensional structure from Nucleotide Sequence Databases nr: GenBank+EMBL+DDBJ+PDB (no EST, STS, GSS, or WGS, or PAT). est: Expressed Seq. tags. 34 billion seq.! htgs: Unfinished High Throughput Genomic Sequences: phases 0, 1 and 2 gss: Genome Survey Sequence,. wgs: Whole Genome Shotgun Sequences. 148 billion sequences
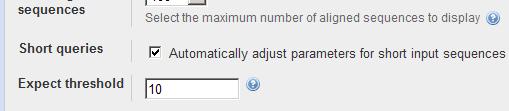
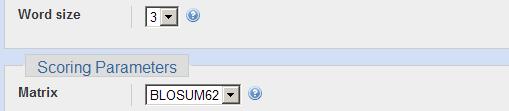
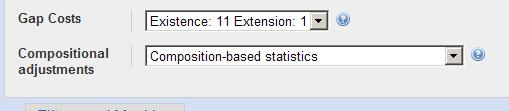
6 BLAST Advanced options -G Cost to open a gap [Integer]; default = 11 ( ) -E Cost to extend a gap [Integer]; default = 1 ( ) -e Expectation value (E) [Real]; default = W Word size; default is 11 for blastn, 3 for other programs. -b Number of alignments to show (B) [Integer]; default = 100 Default Short Query Special Cases Large Sequence Family Ungapped BLAST Filter on off on on Scoring Matrix BLOSUM62 PAM30-35 BLOSUM62 BLOSUM62 Word Size 3 3-2, 7 for DNA 3, 11 for DNA 3, 11 for DNA E value or more Gap costs 11, 1 9, 1 11, 1 4 Alignments





7 Report by species Database: All nr GenBank CDS translations+pdb+swissprot+pir+prf 2,794,673 sequences; 957,836,323 total letters Taxonomy reports Query= Apetala1 P35631 (255 letters + indicates conservative amino acid substitution indicates gap/insertion XXXX shows areas of low complexity CONSIDER TAXONOMIC RELATIONSHIP WHEN INTERPRETING SIMILARITY VALUES!

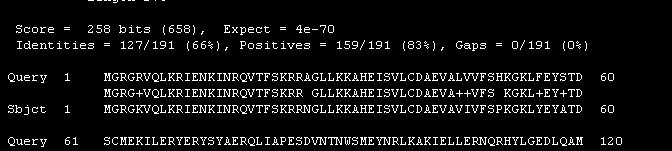
8 Format BLAST output All sequences above the E value threshold are aligned beneath the query. In "with identity identical residues are shown as dots. Flat Query-Anchored Query-Anchored with identities

9 Statistical significance Chance alignments have no biological significance Statistical significance implies low probability of generating a chance alignment Probability of long alignments increases with longer sequences The extreme-value distribution Used to calculate the probability of chance alignment Generated by calculating the scores resulting from repeatedly scrambling one of the sequences being compared
10 BLAST statistics S (Bit score): calculated from raw score S (sum of BLOSUM62 scores) by normalizing with statistical variables that define a scoring system (K and λ). Bit scores from different alignments, even employing different scoring matrices can be compared. S =(λs-lnk)/ln2 k= minor constant λ= constant to adjust for scoring matrix S= score of High-scoring segment pair (HSP) E (expect) value: number of chance alignments with scores equivalent to or better than S that are expected to occur in a database search by chance. E = mn2 -s m= query size N= database size S = bit score m*n= search space The E-value decreases exponentially as the Score (S) that is assigned to a match between two sequences increases. The E-value depends on the size of database and the scoring system in use. When the E-value threshold is increased from the default value of 10, more hits can be reported. When reduced, more significant hits are reported. The lower the E-value (or higher the bit score), the more significant the hit The product mn defines the search space. the same HSP may come out statistically significant in a small database and not significant in a large database
11 P values P: Probability bilit of finding at least one HSP with bit score S or higher by chance. Since it can be shown that t the number of random HSPs with score S' is described by Poisson distribution, the probability of finding at least one HSP with bit score S' is P = 1- e -E E= expect value E= 10 -> P = E= > P =0.01 E= 1 -> P =0.63 E= > P =0.001 E= 0.1 -> P =0.095 E= > P = P-values vary from 0 to 1, whereas E-values can be much greater than 1. The BLAST programs report E-values, rather than P-values, because E-values of, for example, 5 and 10 are much easier to comprehend than P-values of and However, for E < 0.01, P-value and E-value are nearly identical.
12 BLAST Tips Suggested BLAST cutoffs: DNA: book suggests E values < E -6 (I use E<e -10 ) Protein: book suggests E values < E -3 Consider evolutionary divergence in your results!: DNA mutation rate without selection = per site per year. So in 10 million years (10 7 ) of divergences= =0.05 ~ 95% identity BLAST search artifacts: Repeated amino acid stretches (e.g. poly glutamine) or nucleotide repeats (e.g. ATATATATATATAT) result in meaningless positives with significant E values. Use BLAST filters to mask low complexity regions: programs SEG for proteins and DUST for DNA Or customize masking using lower case letter option RepeatMasker can be used to mask repeats in lower case letters
13 MEGABLAST Variation of BLASTN, 10 times faster Optimized for long or highly similar (>95%) sequences Ideal to find whether a large sequence is part of a large contig or chromosome, find sequencing errors and comparing large similar sequences Uses longer default word length (word length= 28 instead of 11) Faster non-affine gap penalty: gap opening penalty=0, gap extension penalty E= r/2 - q (r= match reward Non-affine gapping tends to yield more gaps of shorter length. Accepts multiple consecutive FASTA files as input Discontinuous MEGABLAST q= mismatch penalty) Ideal to compare divergent sequences from different organisms (<80% =) Uses a discontiguous word approach, different from other BLAST programs Nonconsecutive positions are examined over longer segments
 for every position in the alignment.")
 and the results of each iteration used to refine the profile. Each iteration uses a PSSM built from the previous iteration.")
14 PSI-BLAST (Position Specific Iterative BLAST) Designed to detect t weak relationships The added sensitivity comes from the use of a profile that is constructed (automatically) from a multiple alignment. The profile is generated by calculating a Position-Specific Scoring Matrix (PSSM) for every position in the alignment. Also called profiles of Hidden Markov Models PSSM are numerical representations of a multiple alignment A highly conserved ed position receives es a high score. The profile is used to perform additional searches ( iteration) and the results of each iteration used to refine the profile. Each iteration uses a PSSM built from the previous iteration. Continue search iteratively until no new matches are identified: "convergence". Construction of a PSSM PSI-BLAST steps BLASTP Multiple Alignment Construct PSSM Use PSSM to search Each columns in the alignment is a row in the PSSM Frequency of occurrence of a residue at each position Calculate Pb of each aa at each position T at position 8 conserved= highest score 150 P at position 9 less conserve= score 89 Note low scores of aromatic FYW relative to A at P row

15 PHI-BLAST (Pattern Hit Initiated BLAST) PHI-BLAST searches for particular patterns in protein queries. Combines matching of regular expressions with local alignments surrounding the match. PHI-BLAST is preferable to just searching for pattern occurrences because it filters out cases where the pattern occurrence is pb. random and not indicative of homology. PHI-BLAST expects as input a protein query sequence and a pattern contained in that sequence. PHI-BLAST limits alignments to those that match the provided pattern. Statistical significance is reported using E-values as for other forms of BLAST, but the statistical method for computing the E-values is different. PHI-BLAST is integrated with Position-Specific Iterated BLAST (PSI-BLAST), so that the results of aphiblast PHI-BLAST query can be used for PSI-BLAST. Pattern: [C]-x(2)-[C]-x(10,16)-[H]-x(2,3)-[H] Syntax for pattern at
16 Specialized BLAST Great tool! Multiple Sequence Alignment COBALT
Data Retrieval from GenBank
 Data Retrieval from GenBank Peter J. Myler Bioinformatics of Intracellular Pathogens JNU, Feb 7-0, 2009 http://www.ncbi.nlm.nih.gov (January, 2007) http://ncbi.nlm.nih.gov/sitemap/resourceguide.html Accessing
Data Retrieval from GenBank Peter J. Myler Bioinformatics of Intracellular Pathogens JNU, Feb 7-0, 2009 http://www.ncbi.nlm.nih.gov (January, 2007) http://ncbi.nlm.nih.gov/sitemap/resourceguide.html Accessing
CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU
 CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU !2 Sequence Alignment! Global: Needleman-Wunsch-Sellers (1970).! Local: Smith-Waterman (1981) Useful when commonality
CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU !2 Sequence Alignment! Global: Needleman-Wunsch-Sellers (1970).! Local: Smith-Waterman (1981) Useful when commonality
Why learn sequence database searching? Searching Molecular Databases with BLAST
 Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
NCBI Molecular Biology Resources
 NCBI Molecular Biology Resources Part 2: Using NCBI BLAST December 2009 Using BLAST Basics of using NCBI BLAST Using the new Interface Improved organism and filter options New Services Primer BLAST Align
NCBI Molecular Biology Resources Part 2: Using NCBI BLAST December 2009 Using BLAST Basics of using NCBI BLAST Using the new Interface Improved organism and filter options New Services Primer BLAST Align
Match the Hash Scores
 Sort the hash scores of the database sequence February 22, 2001 1 Match the Hash Scores February 22, 2001 2 Lookup method for finding an alignment position 1 2 3 4 5 6 7 8 9 10 11 protein 1 n c s p t a.....
Sort the hash scores of the database sequence February 22, 2001 1 Match the Hash Scores February 22, 2001 2 Lookup method for finding an alignment position 1 2 3 4 5 6 7 8 9 10 11 protein 1 n c s p t a.....
Sequence Based Function Annotation. Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University
 Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
Protein Sequence Analysis. BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl)
") Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
BLAST. Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences.
 BLAST Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences. An example could be aligning an mrna sequence to genomic DNA. Proteins are frequently composed of
BLAST Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences. An example could be aligning an mrna sequence to genomic DNA. Proteins are frequently composed of
Database Searching and BLAST Dannie Durand
 Computational Genomics and Molecular Biology, Fall 2013 1 Database Searching and BLAST Dannie Durand Tuesday, October 8th Review: Karlin-Altschul Statistics Recall that a Maximal Segment Pair (MSP) is
Computational Genomics and Molecular Biology, Fall 2013 1 Database Searching and BLAST Dannie Durand Tuesday, October 8th Review: Karlin-Altschul Statistics Recall that a Maximal Segment Pair (MSP) is
Sequence Based Function Annotation
 Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Sequence Based Function Annotation 1. Given a sequence, how to predict its biological
Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Sequence Based Function Annotation 1. Given a sequence, how to predict its biological
Evolutionary Genetics. LV Lecture with exercises 6KP
 Evolutionary Genetics LV 25600-01 Lecture with exercises 6KP HS2017 >What_is_it? AATGATACGGCGACCACCGAGATCTACACNNNTC GTCGGCAGCGTC 2 NCBI MegaBlast search (09/14) 3 NCBI MegaBlast search (09/14) 4 Submitted
Evolutionary Genetics LV 25600-01 Lecture with exercises 6KP HS2017 >What_is_it? AATGATACGGCGACCACCGAGATCTACACNNNTC GTCGGCAGCGTC 2 NCBI MegaBlast search (09/14) 3 NCBI MegaBlast search (09/14) 4 Submitted
Outline. Evolution. Adaptive convergence. Common similarity problems. Chapter 7: Similarity searches on sequence databases
 Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
A Prac'cal Guide to NCBI BLAST
 A Prac'cal Guide to NCBI BLAST Leonardo Mariño-Ramírez NCBI, NIH Bethesda, USA June 2018 1 NCBI Search Services and Tools Entrez integrated literature and molecular databases Viewers BLink protein similarities
A Prac'cal Guide to NCBI BLAST Leonardo Mariño-Ramírez NCBI, NIH Bethesda, USA June 2018 1 NCBI Search Services and Tools Entrez integrated literature and molecular databases Viewers BLink protein similarities
The String Alignment Problem. Comparative Sequence Sizes. The String Alignment Problem. The String Alignment Problem.
 Dec-82 Oct-84 Aug-86 Jun-88 Apr-90 Feb-92 Nov-93 Sep-95 Jul-97 May-99 Mar-01 Jan-03 Nov-04 Sep-06 Jul-08 May-10 Mar-12 Growth of GenBank 160,000,000,000 180,000,000 Introduction to Bioinformatics Iosif
Dec-82 Oct-84 Aug-86 Jun-88 Apr-90 Feb-92 Nov-93 Sep-95 Jul-97 May-99 Mar-01 Jan-03 Nov-04 Sep-06 Jul-08 May-10 Mar-12 Growth of GenBank 160,000,000,000 180,000,000 Introduction to Bioinformatics Iosif
Creation of a PAM matrix
 Rationale for substitution matrices Substitution matrices are a way of keeping track of the structural, physical and chemical properties of the amino acids in proteins, in such a fashion that less detrimental
Rationale for substitution matrices Substitution matrices are a way of keeping track of the structural, physical and chemical properties of the amino acids in proteins, in such a fashion that less detrimental
Dynamic Programming Algorithms
 Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools
 CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
Introduction to sequence similarity searches and sequence alignment
 Introduction to sequence similarity searches and sequence alignment MBV-INF4410/9410/9410A Monday 18 November 2013 Torbjørn Rognes Department of Informatics, University of Oslo & Department of Microbiology,
Introduction to sequence similarity searches and sequence alignment MBV-INF4410/9410/9410A Monday 18 November 2013 Torbjørn Rognes Department of Informatics, University of Oslo & Department of Microbiology,
Modern BLAST Programs
 Modern BLAST Programs Jian Ma and Louxin Zhang Abstract The Basic Local Alignment Search Tool (BLAST) is arguably the most widely used program in bioinformatics. By sacrificing sensitivity for speed, it
Modern BLAST Programs Jian Ma and Louxin Zhang Abstract The Basic Local Alignment Search Tool (BLAST) is arguably the most widely used program in bioinformatics. By sacrificing sensitivity for speed, it
Basic Local Alignment Search Tool
 14.06.2010 Table of contents 1 History History 2 global local 3 Score functions Score matrices 4 5 Comparison to FASTA References of BLAST History the program was designed by Stephen W. Altschul, Warren
14.06.2010 Table of contents 1 History History 2 global local 3 Score functions Score matrices 4 5 Comparison to FASTA References of BLAST History the program was designed by Stephen W. Altschul, Warren
Textbook Reading Guidelines
 Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Making Sense of DNA and Protein Sequences. Lily Wang, PhD Department of Biostatistics Vanderbilt University
 Making Sense of DNA and Protein Sequences Lily Wang, PhD Department of Biostatistics Vanderbilt University 1 Outline Biological background Major biological sequence databanks Basic concepts in sequence
Making Sense of DNA and Protein Sequences Lily Wang, PhD Department of Biostatistics Vanderbilt University 1 Outline Biological background Major biological sequence databanks Basic concepts in sequence
Question 2: There are 5 retroelements (2 LINEs and 3 LTRs), 6 unclassified elements (XDMR and XDMR_DM), and 7 satellite sequences.
, 6 unclassified elements (XDMR and XDMR_DM), and 7 satellite sequences.") Bio4342 Exercise 1 Answers: Detecting and Interpreting Genetic Homology (Answers prepared by Wilson Leung) Question 1: Low complexity DNA can be described as sequences that consist primarily of one or
Bio4342 Exercise 1 Answers: Detecting and Interpreting Genetic Homology (Answers prepared by Wilson Leung) Question 1: Low complexity DNA can be described as sequences that consist primarily of one or
What I hope you ll learn. Introduction to NCBI & Ensembl tools including BLAST and database searching!
 What I hope you ll learn Introduction to NCBI & Ensembl tools including BLAST and database searching What do we learn from database searching and sequence alignments What tools are available at NCBI What
What I hope you ll learn Introduction to NCBI & Ensembl tools including BLAST and database searching What do we learn from database searching and sequence alignments What tools are available at NCBI What
Alignment to a database. November 3, 2016
 Alignment to a database November 3, 2016 How do you create a database? 1982 GenBank (at LANL, 2000 sequences) 1988 A way to search GenBank (FASTA) Genome Project 1982 GenBank (at LANL, 2000 sequences)
Alignment to a database November 3, 2016 How do you create a database? 1982 GenBank (at LANL, 2000 sequences) 1988 A way to search GenBank (FASTA) Genome Project 1982 GenBank (at LANL, 2000 sequences)
Sequence Analysis. BBSI 2006: Lecture #(χ+3) Takis Benos (2006) BBSI MAY P. Benos 1
 Takis Benos (2006) BBSI MAY P. Benos 1") Sequence Analysis (part III) BBSI 2006: Lecture #(χ+3) Takis Benos (2006) BBSI 2006 31-MAY-2006 2006 P. Benos 1 Outline Sequence variation Distance measures Scoring matrices Pairwise alignments (global,
Sequence Analysis (part III) BBSI 2006: Lecture #(χ+3) Takis Benos (2006) BBSI 2006 31-MAY-2006 2006 P. Benos 1 Outline Sequence variation Distance measures Scoring matrices Pairwise alignments (global,
Genomics I. Organization of the Genome
 Genomics I Organization of the Genome Outline Organization of genome Genomes, chromosomes, genes, exons, introns, promoters, enhancers, etc. Databases Why do we need them? How do we access them? What can
Genomics I Organization of the Genome Outline Organization of genome Genomes, chromosomes, genes, exons, introns, promoters, enhancers, etc. Databases Why do we need them? How do we access them? What can
Comparative Bioinformatics. BSCI348S Fall 2003 Midterm 1
 BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
G4120: Introduction to Computational Biology
 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Lecture 3 February 13, 2003 Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics
G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Lecture 3 February 13, 2003 Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics
BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology
 BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology Jeremy Buhler March 15, 2004 In this lab, we ll annotate an interesting piece of the D. melanogaster genome. Along the way, you ll get
BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology Jeremy Buhler March 15, 2004 In this lab, we ll annotate an interesting piece of the D. melanogaster genome. Along the way, you ll get
UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics
 UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, 2010 SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics DURATION: 3 HOURS TOTAL MARKS: 125 Internal Examiner: Dr. Ché Pillay External Examiner: Prof. Nicola
UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, 2010 SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics DURATION: 3 HOURS TOTAL MARKS: 125 Internal Examiner: Dr. Ché Pillay External Examiner: Prof. Nicola
Optimization of Process Parameters of Global Sequence Alignment Based Dynamic Program - an Approach to Enhance the Sensitivity.
 Optimization of Process Parameters of Global Sequence Alignment Based Dynamic Program - an Approach to Enhance the Sensitivity of Alignment Dr.D.Chandrakala 1, Dr.T.Sathish Kumar 2, S.Preethi 3, D.Sowmya
Optimization of Process Parameters of Global Sequence Alignment Based Dynamic Program - an Approach to Enhance the Sensitivity of Alignment Dr.D.Chandrakala 1, Dr.T.Sathish Kumar 2, S.Preethi 3, D.Sowmya
Typically, to be biologically related means to share a common ancestor. In biology, we call this homologous
 Typically, to be biologically related means to share a common ancestor. In biology, we call this homologous. Two proteins sharing a common ancestor are said to be homologs. Homologyoften implies structural
Typically, to be biologically related means to share a common ancestor. In biology, we call this homologous. Two proteins sharing a common ancestor are said to be homologs. Homologyoften implies structural
Files for this Tutorial: All files needed for this tutorial are compressed into a single archive: [BLAST_Intro.tar.gz]
![Files for this Tutorial: All files needed for this tutorial are compressed into a single archive: [BLAST_Intro.tar.gz]](/thumbs/75/71877651.jpg "Files for this Tutorial: All files needed for this tutorial are compressed into a single archive: [BLAST_Intro.tar.gz]") BLAST Exercise: Detecting and Interpreting Genetic Homology Adapted by W. Leung and SCR Elgin from Detecting and Interpreting Genetic Homology by Dr. J. Buhler Prequisites: None Resources: The BLAST web
BLAST Exercise: Detecting and Interpreting Genetic Homology Adapted by W. Leung and SCR Elgin from Detecting and Interpreting Genetic Homology by Dr. J. Buhler Prequisites: None Resources: The BLAST web
BME 110 Midterm Examination
 BME 110 Midterm Examination May 10, 2011 Name: (please print) Directions: Please circle one answer for each question, unless the question specifies "circle all correct answers". You can use any resource
BME 110 Midterm Examination May 10, 2011 Name: (please print) Directions: Please circle one answer for each question, unless the question specifies "circle all correct answers". You can use any resource
Bioinformatic Methods I Lab 2 LAB 2 ADVANCED BLAST AND COMPARATIVE GENOMICS. [Software needed: web access]
![Bioinformatic Methods I Lab 2 LAB 2 ADVANCED BLAST AND COMPARATIVE GENOMICS. [Software needed: web access]](/thumbs/78/77290464.jpg "Bioinformatic Methods I Lab 2 LAB 2 ADVANCED BLAST AND COMPARATIVE GENOMICS. [Software needed: web access]") LAB 2 ADVANCED BLAST AND COMPARATIVE GENOMICS [Software needed: web access] There are 4 sections to this lab: BlastP, PSI-Blast, Translated Blast, and Comparative Genomics. Last time we used BLAST to query
LAB 2 ADVANCED BLAST AND COMPARATIVE GENOMICS [Software needed: web access] There are 4 sections to this lab: BlastP, PSI-Blast, Translated Blast, and Comparative Genomics. Last time we used BLAST to query
B L A S T! BLAST: Basic local alignment search tool 11/23/2010. Copyright notice. November 29, Outline of today s lecture BLAST. Why use BLAST?
 November 29, 2010 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Bioinformatics pevsner@kennedykrieger.org Johns Hopkins School of Medicine Copyright notice Many of the images
November 29, 2010 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Bioinformatics pevsner@kennedykrieger.org Johns Hopkins School of Medicine Copyright notice Many of the images
Last Update: 12/31/2017. Recommended Background Tutorial: An Introduction to NCBI BLAST
 BLAST Exercise: Detecting and Interpreting Genetic Homology Adapted by T. Cordonnier, C. Shaffer, W. Leung and SCR Elgin from Detecting and Interpreting Genetic Homology by Dr. J. Buhler Recommended Background
BLAST Exercise: Detecting and Interpreting Genetic Homology Adapted by T. Cordonnier, C. Shaffer, W. Leung and SCR Elgin from Detecting and Interpreting Genetic Homology by Dr. J. Buhler Recommended Background
Introduction to Bioinformatics CPSC 265. What is bioinformatics? Textbooks
 Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
Why Use BLAST? David Form - August 15,
 Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
The University of California, Santa Cruz (UCSC) Genome Browser
 Genome Browser") The University of California, Santa Cruz (UCSC) Genome Browser There are hundreds of available userselected tracks in categories such as mapping and sequencing, phenotype and disease associations, genes,
The University of California, Santa Cruz (UCSC) Genome Browser There are hundreds of available userselected tracks in categories such as mapping and sequencing, phenotype and disease associations, genes,
Exercise I, Sequence Analysis
 Exercise I, Sequence Analysis atgcacttgagcagggaagaaatccacaaggactcaccagtctcctggtctgcagagaagacagaatcaacatgagcacagcaggaaaa gtaatcaaatgcaaagcagctgtgctatgggagttaaagaaacccttttccattgaggaggtggaggttgcacctcctaaggcccatgaagt
Exercise I, Sequence Analysis atgcacttgagcagggaagaaatccacaaggactcaccagtctcctggtctgcagagaagacagaatcaacatgagcacagcaggaaaa gtaatcaaatgcaaagcagctgtgctatgggagttaaagaaacccttttccattgaggaggtggaggttgcacctcctaaggcccatgaagt
Sequence Databases and database scanning
 Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Gapped BLAST and PSI-BLAST: a new generation of protein database search programs
 1997 Oxford University Press Nucleic Acids Research, 1997, Vol. 25, No. 17 3389 3402 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs Stephen F. Altschul*, Thomas L. Madden,
1997 Oxford University Press Nucleic Acids Research, 1997, Vol. 25, No. 17 3389 3402 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs Stephen F. Altschul*, Thomas L. Madden,
G4120: Introduction to Computational Biology
 ICB Fall 2009 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Copyright 2009 Oliver Jovanovic, All Rights Reserved. Analysis of Protein
ICB Fall 2009 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Copyright 2009 Oliver Jovanovic, All Rights Reserved. Analysis of Protein
Methods and tools for exploring functional genomics data
 Methods and tools for exploring functional genomics data William Stafford Noble Department of Genome Sciences Department of Computer Science and Engineering University of Washington Outline Searching for
Methods and tools for exploring functional genomics data William Stafford Noble Department of Genome Sciences Department of Computer Science and Engineering University of Washington Outline Searching for
Bioinformatics Tools. Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine
 Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
FUNCTIONAL BIOINFORMATICS
 Molecular Biology-2018 1 FUNCTIONAL BIOINFORMATICS PREDICTING THE FUNCTION OF AN UNKNOWN PROTEIN Suppose you have found the amino acid sequence of an unknown protein and wish to find its potential function.
Molecular Biology-2018 1 FUNCTIONAL BIOINFORMATICS PREDICTING THE FUNCTION OF AN UNKNOWN PROTEIN Suppose you have found the amino acid sequence of an unknown protein and wish to find its potential function.
UCSC Genome Browser. Introduction to ab initio and evidence-based gene finding
 UCSC Genome Browser Introduction to ab initio and evidence-based gene finding Wilson Leung 06/2006 Outline Introduction to annotation ab initio gene finding Basics of the UCSC Browser Evidence-based gene
UCSC Genome Browser Introduction to ab initio and evidence-based gene finding Wilson Leung 06/2006 Outline Introduction to annotation ab initio gene finding Basics of the UCSC Browser Evidence-based gene
ab initio and Evidence-Based Gene Finding
 ab initio and Evidence-Based Gene Finding A basic introduction to annotation Outline What is annotation? ab initio gene finding Genome databases on the web Basics of the UCSC browser Evidence-based gene
ab initio and Evidence-Based Gene Finding A basic introduction to annotation Outline What is annotation? ab initio gene finding Genome databases on the web Basics of the UCSC browser Evidence-based gene
Application for Automating Database Storage of EST to Blast Results. Vikas Sharma Shrividya Shivkumar Nathan Helmick
 Application for Automating Database Storage of EST to Blast Results Vikas Sharma Shrividya Shivkumar Nathan Helmick Outline Biology Primer Vikas Sharma System Overview Nathan Helmick Creating ESTs Nathan
Application for Automating Database Storage of EST to Blast Results Vikas Sharma Shrividya Shivkumar Nathan Helmick Outline Biology Primer Vikas Sharma System Overview Nathan Helmick Creating ESTs Nathan
ELE4120 Bioinformatics. Tutorial 5
 ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
Protein Bioinformatics Part I: Access to information
 Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Comparative Genomics. Page 1. REMINDER: BMI 214 Industry Night. We ve already done some comparative genomics. Loose Definition. Human vs.
 Page 1 REMINDER: BMI 214 Industry Night Comparative Genomics Russ B. Altman BMI 214 CS 274 Location: Here (Thornton 102), on TV too. Time: 7:30-9:00 PM (May 21, 2002) Speakers: Francisco De La Vega, Applied
Page 1 REMINDER: BMI 214 Industry Night Comparative Genomics Russ B. Altman BMI 214 CS 274 Location: Here (Thornton 102), on TV too. Time: 7:30-9:00 PM (May 21, 2002) Speakers: Francisco De La Vega, Applied
03-511/711 Computational Genomics and Molecular Biology, Fall
 03-511/711 Computational Genomics and Molecular Biology, Fall 2010 1 Study questions These study problems are intended to help you to review for the final exam. This is not an exhaustive list of the topics
03-511/711 Computational Genomics and Molecular Biology, Fall 2010 1 Study questions These study problems are intended to help you to review for the final exam. This is not an exhaustive list of the topics
Annotation of contig27 in the Muller F Element of D. elegans. Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans.
 David Wang Bio 434W 4/27/15 Annotation of contig27 in the Muller F Element of D. elegans Abstract Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans. Genscan predicted six
David Wang Bio 434W 4/27/15 Annotation of contig27 in the Muller F Element of D. elegans Abstract Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans. Genscan predicted six
Single alignment: FASTA. 17 march 2017
 Single alignment: FASTA 17 march 2017 FASTA is a DNA and protein sequence alignment software package first described (as FASTP) by David J. Lipman and William R. Pearson in 1985.[1] FASTA is pronounced
Single alignment: FASTA 17 march 2017 FASTA is a DNA and protein sequence alignment software package first described (as FASTP) by David J. Lipman and William R. Pearson in 1985.[1] FASTA is pronounced
3D Structure Prediction with Fold Recognition/Threading. Michael Tress CNB-CSIC, Madrid
 3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
Data Mining for Biological Data Analysis
 Data Mining for Biological Data Analysis Data Mining and Text Mining (UIC 583 @ Politecnico di Milano) References Data Mining Course by Gregory-Platesky Shapiro available at www.kdnuggets.com Jiawei Han
Data Mining for Biological Data Analysis Data Mining and Text Mining (UIC 583 @ Politecnico di Milano) References Data Mining Course by Gregory-Platesky Shapiro available at www.kdnuggets.com Jiawei Han
Bioinformatic analysis of similarity to allergens. Mgr. Jan Pačes, Ph.D. Institute of Molecular Genetics, Academy of Sciences, CR
 Bioinformatic analysis of similarity to allergens Mgr. Jan Pačes, Ph.D. Institute of Molecular Genetics, Academy of Sciences, CR Scope of the work Method for allergenicity search used by FAO/WHO Analysis
Bioinformatic analysis of similarity to allergens Mgr. Jan Pačes, Ph.D. Institute of Molecular Genetics, Academy of Sciences, CR Scope of the work Method for allergenicity search used by FAO/WHO Analysis
Basic Bioinformatics: Homology, Sequence Alignment,
 Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
03-511/711 Computational Genomics and Molecular Biology, Fall
 03-511/711 Computational Genomics and Molecular Biology, Fall 2011 1 Study questions These study problems are intended to help you to review for the final exam. This is not an exhaustive list of the topics
03-511/711 Computational Genomics and Molecular Biology, Fall 2011 1 Study questions These study problems are intended to help you to review for the final exam. This is not an exhaustive list of the topics
Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 2. Bioinformatics 1: Biology, Sequences, Phylogenetics
 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 2 Sepp Hochreiter gene Central Dogma nucleus DNA 1. transcription (mrna) 2. transport mrna protein 3. translation (ribosom, trna) 4. folding (protein)
Bioinformatics 1 Biology, Sequences, Phylogenetics Part 2 Sepp Hochreiter gene Central Dogma nucleus DNA 1. transcription (mrna) 2. transport mrna protein 3. translation (ribosom, trna) 4. folding (protein)
G4120: Introduction to Computational Biology
 ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
Scoring Alignments. Genome 373 Genomic Informatics Elhanan Borenstein
 Scoring Alignments Genome 373 Genomic Informatics Elhanan Borenstein A quick review Course logistics Genomes (so many genomes) The computational bottleneck Python: Programs, input and output Number and
Scoring Alignments Genome 373 Genomic Informatics Elhanan Borenstein A quick review Course logistics Genomes (so many genomes) The computational bottleneck Python: Programs, input and output Number and
An introduction to multiple alignments
 An introduction to multiple alignments original version by Cédric Notredame, updated by Laurent Falquet Overview! Multiple alignments! How-to, Goal, problems, use! Patterns! PROSITE database, syntax, use!
An introduction to multiple alignments original version by Cédric Notredame, updated by Laurent Falquet Overview! Multiple alignments! How-to, Goal, problems, use! Patterns! PROSITE database, syntax, use!
COMPUTER RESOURCES II:
 COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
Sequence Analysis. Introduction to Bioinformatics BIMMS December 2015
 Sequence Analysis Introduction to Bioinformatics BIMMS December 2015 abriel Teku Department of Experimental Medical Science Faculty of Medicine Lund University Sequence analysis Part 1 Sequence analysis:
Sequence Analysis Introduction to Bioinformatics BIMMS December 2015 abriel Teku Department of Experimental Medical Science Faculty of Medicine Lund University Sequence analysis Part 1 Sequence analysis:
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. Evidence Based Annotation. GEP goals: Evidence for Gene Models 08/22/2017
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. GEP goals: Evidence Based Annotation. Evidence for Gene Models 12/26/2018
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Tutorial for Stop codon reassignment in the wild
 Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Biotechnology Explorer
 Biotechnology Explorer C. elegans Behavior Kit Bioinformatics Supplement explorer.bio-rad.com Catalog #166-5120EDU This kit contains temperature-sensitive reagents. Open immediately and see individual
Biotechnology Explorer C. elegans Behavior Kit Bioinformatics Supplement explorer.bio-rad.com Catalog #166-5120EDU This kit contains temperature-sensitive reagents. Open immediately and see individual
Agenda. Web Databases for Drosophila. Gene annotation workflow. GEP Drosophila annotation projects 01/01/2018. Annotation adding labels to a sequence
 Agenda GEP annotation project overview Web Databases for Drosophila An introduction to web tools, databases and NCBI BLAST Web databases for Drosophila annotation UCSC Genome Browser NCBI / BLAST FlyBase
Agenda GEP annotation project overview Web Databases for Drosophila An introduction to web tools, databases and NCBI BLAST Web databases for Drosophila annotation UCSC Genome Browser NCBI / BLAST FlyBase
DNAFSMiner: A Web-Based Software Toolbox to Recognize Two Types of Functional Sites in DNA Sequences
 DNAFSMiner: A Web-Based Software Toolbox to Recognize Two Types of Functional Sites in DNA Sequences Huiqing Liu Hao Han Jinyan Li Limsoon Wong Institute for Infocomm Research, 21 Heng Mui Keng Terrace,
DNAFSMiner: A Web-Based Software Toolbox to Recognize Two Types of Functional Sites in DNA Sequences Huiqing Liu Hao Han Jinyan Li Limsoon Wong Institute for Infocomm Research, 21 Heng Mui Keng Terrace,
Identifying Genes and Pseudogenes in a Chimpanzee Sequence Adapted from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. M.
 Identifying Genes and Pseudogenes in a Chimpanzee Sequence Adapted from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. M. Brent Prerequisites: A Simple Introduction to NCBI BLAST Resources: The GENSCAN
Identifying Genes and Pseudogenes in a Chimpanzee Sequence Adapted from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. M. Brent Prerequisites: A Simple Introduction to NCBI BLAST Resources: The GENSCAN
Chimp Sequence Annotation: Region 2_3
 Chimp Sequence Annotation: Region 2_3 Jeff Howenstein March 30, 2007 BIO434W Genomics 1 Introduction We received region 2_3 of the ChimpChunk sequence, and the first step we performed was to run RepeatMasker
Chimp Sequence Annotation: Region 2_3 Jeff Howenstein March 30, 2007 BIO434W Genomics 1 Introduction We received region 2_3 of the ChimpChunk sequence, and the first step we performed was to run RepeatMasker
Challenging algorithms in bioinformatics
 Challenging algorithms in bioinformatics 11 October 2018 Torbjørn Rognes Department of Informatics, UiO torognes@ifi.uio.no What is bioinformatics? Definition: Bioinformatics is the development and use
Challenging algorithms in bioinformatics 11 October 2018 Torbjørn Rognes Department of Informatics, UiO torognes@ifi.uio.no What is bioinformatics? Definition: Bioinformatics is the development and use
Why study sequence similarity?
 Sequence Similarity Why study sequence similarity? Possible indication of common ancestry Similarity of structure implies similar biological function even among apparently distant organisms Example context:
Sequence Similarity Why study sequence similarity? Possible indication of common ancestry Similarity of structure implies similar biological function even among apparently distant organisms Example context:
BLAST. Subject: The result from another organism that your query was matched to.
 BLAST (Basic Local Alignment Search Tool) Note: This is a complete transcript to the powerpoint. It is good to read through this once to understand everything. If you ever need help and just need a quick
BLAST (Basic Local Alignment Search Tool) Note: This is a complete transcript to the powerpoint. It is good to read through this once to understand everything. If you ever need help and just need a quick
Bioinformatics & Protein Structural Analysis. Bioinformatics & Protein Structural Analysis. Learning Objective. Proteomics
 The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
Assemblytics: a web analytics tool for the detection of assembly-based variants Maria Nattestad and Michael C. Schatz
 Assemblytics: a web analytics tool for the detection of assembly-based variants Maria Nattestad and Michael C. Schatz Table of Contents Supplementary Note 1: Unique Anchor Filtering Supplementary Figure
Assemblytics: a web analytics tool for the detection of assembly-based variants Maria Nattestad and Michael C. Schatz Table of Contents Supplementary Note 1: Unique Anchor Filtering Supplementary Figure
Two Mark question and Answers
 1. Define Bioinformatics Two Mark question and Answers Bioinformatics is the field of science in which biology, computer science, and information technology merge into a single discipline. There are three
1. Define Bioinformatics Two Mark question and Answers Bioinformatics is the field of science in which biology, computer science, and information technology merge into a single discipline. There are three
Computational Molecular Biology. Lecture Notes. by A.P. Gultyaev
 Computational Molecular Biology Lecture Notes by A.P. Gultyaev Leiden Institute of Applied Computer Science (LIACS) Leiden University January 2017 1 Contents Introduction... 3 1. Sequence databases...
Computational Molecular Biology Lecture Notes by A.P. Gultyaev Leiden Institute of Applied Computer Science (LIACS) Leiden University January 2017 1 Contents Introduction... 3 1. Sequence databases...
Sequence Analysis. II: Sequence Patterns and Matrices. George Bell, Ph.D. WIBR Bioinformatics and Research Computing
 Sequence Analysis II: Sequence Patterns and Matrices George Bell, Ph.D. WIBR Bioinformatics and Research Computing Sequence Patterns and Matrices Multiple sequence alignments Sequence patterns Sequence
Sequence Analysis II: Sequence Patterns and Matrices George Bell, Ph.D. WIBR Bioinformatics and Research Computing Sequence Patterns and Matrices Multiple sequence alignments Sequence patterns Sequence
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Mittwoch
 WS2013/2014 Woche 3 - Mittwoch") VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Mittwoch Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Mittwoch Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
Lecture 17: Heuris.c methods for sequence alignment: BLAST and FASTA. Spring 2017 April 11, 2017
 Lecture 17: Heuris.c methods for sequence alignment: BLAST and FASTA Spring 2017 April 11, 2017 Mo.va.on Smith- Waterman algorithm too slow for searching large sequence databases Most sequences are not
Lecture 17: Heuris.c methods for sequence alignment: BLAST and FASTA Spring 2017 April 11, 2017 Mo.va.on Smith- Waterman algorithm too slow for searching large sequence databases Most sequences are not
Databases in genomics
 Databases in genomics Search in biological databases: The most common task of molecular biologist researcher, to answer to the following ques7ons:! Are they new sequences deposited in biological databases
Databases in genomics Search in biological databases: The most common task of molecular biologist researcher, to answer to the following ques7ons:! Are they new sequences deposited in biological databases
Annotating 7G24-63 Justin Richner May 4, Figure 1: Map of my sequence
 Annotating 7G24-63 Justin Richner May 4, 2005 Zfh2 exons Thd1 exons Pur-alpha exons 0 40 kb 8 = 1 kb = LINE, Penelope = DNA/Transib, Transib1 = DINE = Novel Repeat = LTR/PAO, Diver2 I = LTR/Gypsy, Invader
Annotating 7G24-63 Justin Richner May 4, 2005 Zfh2 exons Thd1 exons Pur-alpha exons 0 40 kb 8 = 1 kb = LINE, Penelope = DNA/Transib, Transib1 = DINE = Novel Repeat = LTR/PAO, Diver2 I = LTR/Gypsy, Invader
MATH 5610, Computational Biology
 MATH 5610, Computational Biology Lecture 2 Intro to Molecular Biology (cont) Stephen Billups University of Colorado at Denver MATH 5610, Computational Biology p.1/24 Announcements Error on syllabus Class
MATH 5610, Computational Biology Lecture 2 Intro to Molecular Biology (cont) Stephen Billups University of Colorado at Denver MATH 5610, Computational Biology p.1/24 Announcements Error on syllabus Class
Annotating Fosmid 14p24 of D. Virilis chromosome 4
 Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
Chimp BAC analysis: Adapted by Wilson Leung and Sarah C.R. Elgin from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. Michael R.
 Chimp BAC analysis: Adapted by Wilson Leung and Sarah C.R. Elgin from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. Michael R. Brent Prerequisites: BLAST exercise: Detecting and Interpreting Genetic
Chimp BAC analysis: Adapted by Wilson Leung and Sarah C.R. Elgin from Chimp BAC analysis: TWINSCAN and UCSC Browser by Dr. Michael R. Brent Prerequisites: BLAST exercise: Detecting and Interpreting Genetic
Theory and Application of Multiple Sequence Alignments
 Theory and Application of Multiple Sequence Alignments a.k.a What is a Multiple Sequence Alignment, How to Make One, and What to Do With It Brett Pickett, PhD History Structure of DNA discovered (1953)
Theory and Application of Multiple Sequence Alignments a.k.a What is a Multiple Sequence Alignment, How to Make One, and What to Do With It Brett Pickett, PhD History Structure of DNA discovered (1953)
BIOINFORMATICS TO ANALYZE AND COMPARE GENOMES
 BIOINFORMATICS TO ANALYZE AND COMPARE GENOMES We sequenced and assembled a genome, but this is only a long stretch of ATCG What should we do now? 1. find genes What are the starting and end points for
BIOINFORMATICS TO ANALYZE AND COMPARE GENOMES We sequenced and assembled a genome, but this is only a long stretch of ATCG What should we do now? 1. find genes What are the starting and end points for
Imaging informatics computer assisted mammogram reading Clinical aka medical informatics CDSS combining bioinformatics for diagnosis, personalized
 1 2 3 Imaging informatics computer assisted mammogram reading Clinical aka medical informatics CDSS combining bioinformatics for diagnosis, personalized medicine, risk assessment etc Public Health Bio
1 2 3 Imaging informatics computer assisted mammogram reading Clinical aka medical informatics CDSS combining bioinformatics for diagnosis, personalized medicine, risk assessment etc Public Health Bio
BLASTing through the kingdom of life
 Information for teachers Description: In this activity, students copy unknown DNA sequences and use them to search GenBank, the main database of nucleotide sequences at the National Center for Biotechnology
Information for teachers Description: In this activity, students copy unknown DNA sequences and use them to search GenBank, the main database of nucleotide sequences at the National Center for Biotechnology
Protein Structure Prediction. christian studer , EPFL
 Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
Gene Annotation Project. Group 1. Tyler Tiede Yanzhu Ji Jenae Skelton
 Gene Annotation Project Group 1 Tyler Tiede Yanzhu Ji Jenae Skelton Outline Tools Overview of 150kb region Overview of annotation process Characterization of 5 putative gene regions Analysis of masked
Gene Annotation Project Group 1 Tyler Tiede Yanzhu Ji Jenae Skelton Outline Tools Overview of 150kb region Overview of annotation process Characterization of 5 putative gene regions Analysis of masked
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part II. Protein Domain Identification & Classification Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part II. Protein Domain Identification & Classification Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
A History of Bioinformatics: Development of in silico Approaches to Evaluate Food Proteins
 A History of Bioinformatics: Development of in silico Approaches to Evaluate Food Proteins /////////// Andre Silvanovich Ph. D. Bayer Crop Sciences Chesterfield, MO October 2018 Bioinformatic Evaluation
A History of Bioinformatics: Development of in silico Approaches to Evaluate Food Proteins /////////// Andre Silvanovich Ph. D. Bayer Crop Sciences Chesterfield, MO October 2018 Bioinformatic Evaluation
From assembled genome to annotated genome
 From assembled genome to annotated genome Procaryotic genomes Eucaryotic genomes Genome annotation servers (web based) 1. RAST 2. NCBI Gene prediction pipeline: Maker Function annotation pipeline: Blast2GO
From assembled genome to annotated genome Procaryotic genomes Eucaryotic genomes Genome annotation servers (web based) 1. RAST 2. NCBI Gene prediction pipeline: Maker Function annotation pipeline: Blast2GO