10/06/2014. RNA-Seq analysis. With reference assembly. Cormier Alexandre, PhD student UMR8227, Algal Genetics Group
|
|
|
- Vanessa Quinn
- 5 years ago
- Views:
Transcription







1 RNA-Seq analysis With reference assembly Cormier Alexandre, PhD student UMR8227, Algal Genetics Group
2 Summary 2
3 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference genome No reference transcriptome 3
4 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference genome No reference transcriptome Non discovery mode RNA-seq reads QC + Cleaning Mapping Differential Expression Analysis 4


5 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference genome No reference transcriptome Non discovery mode Discovery mode RNA-seq reads RNA-seq reads QC + Cleaning QC + Cleaning Mapping Mapping Assembly Differential Expression Analysis Differential Expression Analysis 5


6 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference genome No reference transcriptome Non discovery mode Discovery mode RNA-seq reads RNA-seq reads RNA-seq reads QC + Cleaning QC + Cleaning QC + Cleaning Mapping Mapping Mapping Assembly Assembly Differential Expression Analysis Differential Expression Analysis Differential Expression Analysis 6
7 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference genome No reference transcriptome Non discovery mode Discovery mode RNA-seq reads RNA-seq reads RNA-seq reads QC + Cleaning QC + Cleaning QC + Cleaning Mapping Mapping Mapping Assembly Assembly Differential Expression Analysis Differential Expression Analysis Differential Expression Analysis 7
8 Tuxedo Workflow v2.2.0 Introduction 8
9 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped 9
10 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts 10
11 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Final transcriptome assembly 11
12 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Mapped Final transcriptome assembly Mapped Cuffquant 12
13 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Mapped Final transcriptome assembly Mapped Cuffquant Cuffdiff Differential expression results CummeRbund Expression plots 13
14 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Mapped Final transcriptome assembly Mapped Cuffquant Cuffdiff Cuffnorm Differential expression results Normalized expression & count tables CummeRbund R, Matlab, etc Expression plots 14
15 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Mapped Final transcriptome assembly Mapped Mapped Mapped Cuffquant HTSeq Cuffdiff Cuffnorm Gene quantification Differential expression results Normalized expression & count tables DESeq/EdgeR CummeRbund R, Matlab, etc Differential expression results Expression plots 15
16 Tuxedo Workflow v2.2.0 Introduction Bowtie Condition A Condition B TopHat Mapped Mapped Cufflinks Assembled transcripts Assembled transcripts Cuffmerge Cuffcompare Compare to reference annotation Mapped Mapped HTSeq Gene quantification DESeq/EdgeR Differential expression results 16
17 Other solutions Introduction Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 17
18 Data presentation Data Data retrived from the ENCODE project 2 human cell lines : Gm12878 (lymphoblastoid cell line) 2 replicates Hct116 (colorectal carcinoma cell line) 2 replicates Illumina paired-end 2x75bp, insert size ~400bp Working only on the chromosome 22 Objective : Identify differentially expressed genes in 2 human cell lines 18


19 Get data Data 19

20 Get data Data Export all data in a new history and choose a name (ex: rna-seq reference analysis) 20

21 Data exploration: FastQC Quality control Obtain some statistics and information of a fastq file Check the quality of the data contained in fastq file 21
22 Data exploration: FastQC Quality control Launch FastQC analysis only on : Gm12878_rep1_R1.fastq Hct116_rep1_R1.fastq 22

23 Data exploration: FastQC Quality control 23
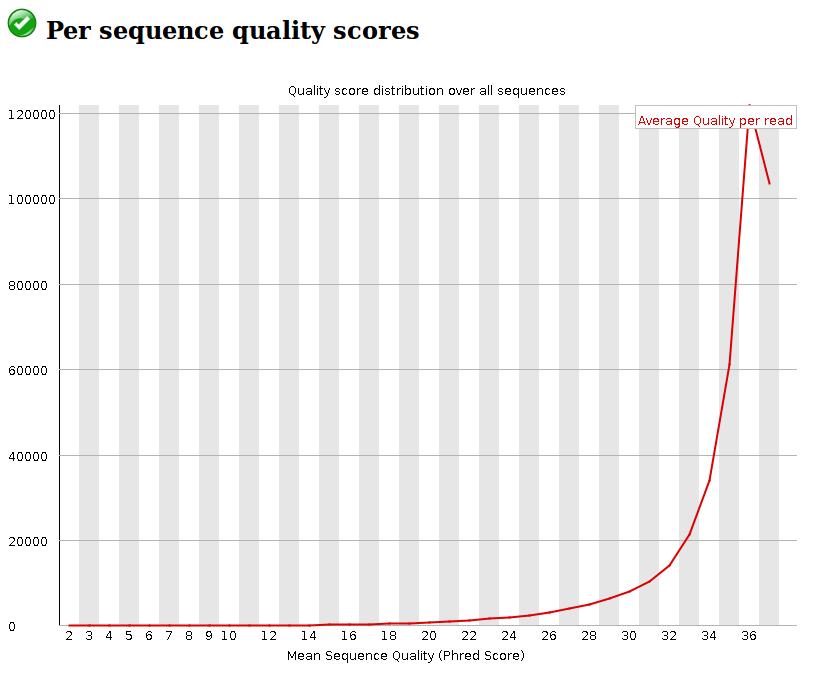



24 Data exploration: FastQC Quality control 24

25 Data exploration: FastQC Quality control 25
26 Cleaning Cleaning with PRINSEQ 26
27 Cleaning: PRINSEQ Cleaning With a reference genome, the cleaning step is not necessary. The use of genome allows filtering reads with a poor quality and contamination. Can be problematic with Illumina reads diminution of the quality at the end of the sequence 27
28 Cleaning: PRINSEQ Cleaning Raw read Mapped? High quality Low quality 28
29 Cleaning: PRINSEQ Cleaning Raw read Cleaned read Mapped? High quality Low quality 29

30 Cleaning: PRINSEQ Cleaning 30
31 PRINSEQ Parameters Cleaning Launch PRINSEQ on all fastq files 31
32 Cleaning: PRINSEQ Cleaning Launch FastQC analysis only on : Gm12878_rep1_R1.fastq_good.fastqsanger Hct116_rep1_R1.fastq_good.fastqsanger Compare results with raw reads 32
33 Mapping Mapping with TopHat 2 33

34 Mapping: TopHat 2 Mapping TopHat is a fast splice junction mapper for RNA-Seq reads. It aligns RNA-Seq reads to genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify splice junctions between exons 34
35 Overview Mapping Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 35
36 Overview Mapping Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 36
37 Overview Mapping Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 37
38 Overview Mapping Faster (~x8) and less greedy Better for polymorphic species A little bit more exhaustive Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 38
39 Overview Mapping Faster (~x8) and less greedy Better for polymorphic species A little bit more exhaustive Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 39
40 TopHat2 Input Mapping Fastq file(s) Genome One mapping per replicate 40

41 TopHat2 Parameters Mapping 1: By default: 20 41

42 Insert size Mapping 42


43 Default parameters Mapping TopHat 2 is optimized for: Human Mouse If you work on these species, you can use default parameters Else, you need to input all of the specie specifics parameters, such as intron size. 43

44 Multiple mapping reads Mapping Some reads will align to more than one place in the reference, because: Shared exons (if reference is transcriptome) Common domains, gene families Paralogs, pseudogenes, etc. This can distort counts, leading to misleading expression levels If a read can t be uniquely mapped, how should it be counted or ignored? Should it be randomly assigned to one location among all the locations to which it aligns equally well? This may depend on the question you re asking......also depends on the software you use and also depends of your data (read length, quality, etc) 44


45 TopHat2 Output Mapping BAM: compressed binary version of the SAM BAM to SAM 45

46 TopHat2 Output Mapping SAM file 46
47 SAM (Sequence Alignment/Map) SN:sctg_997 SN:sctg_998 SN:sctg_999 ID:TopHat VN:2.0.3 HWI-ST132_0435:3:63:3889:100528#GATCAG 0 sctg_ M * 0 0 Reference sequence dictionary Program Sequence ID Flag Reference sequence name Leftmost mapping position Mapping quality CIGAR string Reference of the mate/next read Mapping position of mate/next read Insert size CCCGCCGCTCCATGATCTCCAAGAGGCGCAGCTCTCGCAAGGCTTCCGCCAAGGTGGTGGCTT gggggggggggggggggggggggggggggggeeeggeeggyb^ce^bbbc_cac[ddacaa_c AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:73 YT:Z:UU XS:A:+ NH:i:2 CC:Z:sctg_861 CP:i:9032 HI:i:0 Sequence ID Quality Mapper tag 47
48 SAM flags Mapping 48
49 Assembly Transcripts assembly with Cufflinks 2 49

50 Assembly: Cufflinks Assembly Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA-Seq samples. 50
. Garber, M., Grabherr, M. G., Guttman, M.")
51 Assembly: Cufflinks Assembly Scripture Sensitivity Cufflinks Precision Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech 28, (2010). Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth 8, (2011). 51
52 Cufflinks Input Cufflinks BAM Genome Annotations One assembly per replicate in case of DE analysis 52

53 Cufflinks Parameters Cufflinks 53
54 Why use annotation? Assembly RABT: Reference Annotation Based Transcript assembly Spliced reads (dashed line) Read pairs (solid line) Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics btr355 (2011). doi: /bioinformatics/btr355 54
55 Why use annotation? Assembly RABT: Reference Annotation Based Transcript assembly Spliced reads (dashed line) Read pairs (solid line) Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics btr355 (2011). doi: /bioinformatics/btr355 55
56 Why use annotation? Assembly RABT: Reference Annotation Based Transcript assembly Spliced reads (dashed line) Read pairs (solid line) Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics btr355 (2011). doi: /bioinformatics/btr355 56
. doi:10.")
57 Why use annotation? Assembly RABT: Reference Annotation Based Transcript assembly Spliced reads (dashed line) Read pairs (solid line) Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics btr355 (2011). doi: /bioinformatics/btr355 57
58 Why use annotation? Assembly RABT: Reference Annotation Based Transcript assembly Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics btr355 (2011). doi: /bioinformatics/btr355 58
59 gtf/gff format Assembly GFF (general feature format) is a file format used for describing genes and other features of DNA, RNA and protein sequences. gff3 Seqname Source Score Strand Frame chr22 protein_coding gene ID=ENSG ;Name=SEPT5 chr22 protein_coding mrna ID=ENST ;Name=SEPT5-016;Parent=ENSG chr22 protein_coding protein ID=ENSP ;Name=SEPT5-016;Parent=ENST chr22 protein_coding CDS Name=CDS:SEPT5;Parent=ENST chr22 protein_coding CDS Name=CDS:SEPT5;Parent=ENST chr22 protein_coding CDS Name=CDS:SEPT5;Parent=ENST chr22 protein_coding CDS Name=CDS:SEPT5;Parent=ENST chr22 protein_coding exon Parent=ENST chr22 protein_coding exon Parent=ENST chr22 protein_coding exon Parent=ENST chr22 protein_coding exon Parent=ENST Feature Start End Attribute 59
60 Cufflinks Output Assembly GTF file (x4) Seqname Source Feature Start End Score Strand Frame Attributes chr22 Cufflinks transcript gene_id "CUFF.1"; transcript_id "CUFF.1.1"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; full_read_support "yes"; chr22 Cufflinks exon gene_id "CUFF.1"; transcript_id "CUFF.1.1"; exon_number "1"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; chr22 Cufflinks transcript gene_id "NM_ "; transcript_id "NM_ "; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; full_read_support "no"; chr22 Cufflinks exon gene_id "NM_ "; transcript_id "NM_ "; exon_number "1"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; chr22 Cufflinks exon gene_id "NM_ "; transcript_id "NM_ "; exon_number "2"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; chr22 Cufflinks exon gene_id "NM_ "; transcript_id "NM_ "; exon_number "3"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; chr22 Cufflinks exon gene_id "NM_ "; transcript_id "NM_ "; exon_number "4"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; chr22 Cufflinks exon gene_id "NM_ "; transcript_id "NM_ "; exon_number "5"; FPKM " "; frac " "; conf_lo " "; conf_hi " "; cov " "; 60
 Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5, 621 628 (2008).")
61 FPKM (RPKM) Assembly Fragments Per Kilobase of exon model per Million mapped fragments FPKM 9 10 C NL C= the number of reads mapped onto the gene's exons N= total number of mapped reads L= the sum of the exons in base pairs (transcript length) Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5, (2008). Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech 28, (2010). 61

62 Assembly: Cuffmerge Assembly Cuffmerge is used to merge together several Cufflinks assemblies. It also handles running Cuffcompare for you, and automatically filters a number of transfrags that are probably artifacts. 62
63 Cuffmerge Input Assembly gtf from Cufflinks Genome Annotation 63
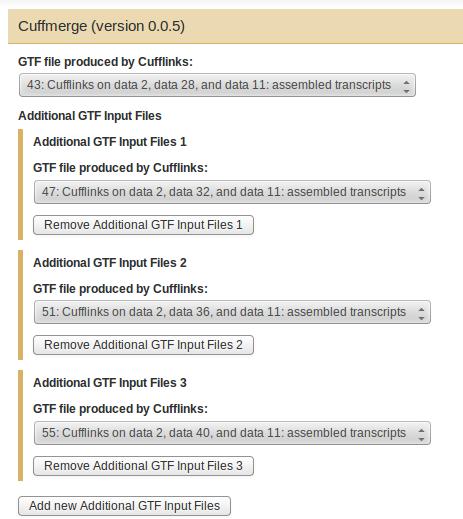

64 Cuffmerge Parameters Assembly 64
65 Cuffmerge Output Assembly gtf (x1) Seqname Source Feature Start End Score Strand Frame Attributes chr22 Cufflinks exon gene_id "XLOC_000001"; transcript_id "TCONS_ "; exon_number "1"; gene_name "NR_073460"; oid "NR_073459"; nearest_ref "NR_073460"; class_code "="; tss_id "TSS1"; chr22 Cufflinks exon gene_id "XLOC_000001"; transcript_id "TCONS_ "; exon_number "2"; gene_name "NR_073460"; oid "NR_073459"; nearest_ref "NR_073460"; class_code "="; tss_id "TSS1"; chr22 Cufflinks exon gene_id "XLOC_000001"; transcript_id "TCONS_ "; exon_number "3"; gene_name "NR_073460"; oid "NR_073459"; nearest_ref "NR_073460"; class_code "="; tss_id "TSS1"; chr22 Cufflinks exon gene_id "XLOC_000002"; transcript_id "TCONS_ "; exon_number "1"; oid "CUFF.3.1"; class_code "u"; tss_id "TSS2"; chr22 Cufflinks exon gene_id "XLOC_000002"; transcript_id "TCONS_ "; exon_number "1"; oid "CUFF.4.1"; class_code "u"; tss_id "TSS2"; chr22 Cufflinks exon gene_id "XLOC_000002"; transcript_id "TCONS_ "; exon_number "2"; oid "CUFF.4.1"; class_code "u"; tss_id "TSS2"; chr22 Cufflinks exon gene_id "XLOC_000003"; transcript_id "TCONS_ "; exon_number "1"; gene_name "NR_001591"; oid "NR_001591"; nearest_ref "NR_001591"; class_code "="; tss_id "TSS3"; chr22 Cufflinks exon gene_id "XLOC_000003"; transcript_id "TCONS_ "; exon_number "2"; gene_name "NR_001591"; oid "NR_001591"; nearest_ref "NR_001591"; class_code "="; tss_id "TSS3"; chr22 Cufflinks exon gene_id "XLOC_000003"; transcript_id "TCONS_ "; exon_number "3"; gene_name "NR_001591"; oid "NR_001591"; nearest_ref "NR_001591"; class_code "="; tss_id "TSS3"; 65

66 Assembly: Cuffcompare Assembly Cuffcompare is used to compare assembled transcripts to a reference annotation. 66
67 Cuffcompare Input Assembly gtf from Cufflinks / Cuffmerge Reference annotation Genome 67
68 Cuffcompare Parameters Assembly 68
69 Cuffcompare Output Assembly The following table shows the code used by Cufflinks to classify the transcripts in comparison with the reference annotation Priority Code Description 1 = Complete match of intron chain 2 c Contained 3 j Potentially novel isoform (fragment): at least one splice junction is shared with a reference transcript 4 e Single exon transfrag overlapping a reference exon and at least 10 bp of a reference intron, indicating a possible pre-mrna fragment 5 i A transfrag falling entirely within a reference intron 6 o Generic exonic overlap with a reference transcript 7 p Possible polymerase run-on fragment (within 2Kbases of a reference transcript) 8 r Repeat. Currently determined by looking at the soft-masked reference sequence and applied to transcripts where at least 50% of the bases are lower case 9 u Unknown, intergenic transcript 10 x Exonic overlap with reference on the opposite strand 11 s An intron of the transfrag overlaps a reference intron on the opposite strand (likely due to read mapping errors) 12. (.tracking file only, indicates multiple classifications) 69

70 Examples Assembly = 70
71 Examples Assembly J 71
72 Examples Assembly U 72
73 Counting Read counting per gene with HTSeq-count 73

74 Counting: HTSeq Counting HTSeq is a Python package that provides infrastructure to process data from high-throughput sequencing assays. 74
75 HTSeq Input Counting BAM gtf/gtf annotation file One counting per replicate 75

76 HTSeq Parameters Counting 76


77 HTSeq Mode Counting 77
78 HTSeq Attribute Counting chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene CDS gene_id "NM_ "; transcript_id "NM_ "; chr22 hg19_refgene exon gene_id "NM_ "; transcript_id "NM_ "; Feature Attribute 78
79 HTSeq Output Counting Tabular file (x4) gene ID Read count NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_ NM_

80 Merging tabular Counting 80
81 Merging tabular Parameters Counting 81
82 Merging tabular Output Counting A matrix Gm12878_1 Gm12878_2 Hct116_1 Hct116_2 NM_ NM_ NM_ NM_ NR_ NR_ NR_ NM_ NM_ NR_ NM_ NM_ NM_ NM_ NM_ NM_ NR_ NM_ NM_ NR_ NR_ NM_ NM_ NM_

83 End 83
VM origin. Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE
 X2go: LXDE") VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
RNA Seq: Methods and Applica6ons. Prat Thiru
 RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA-Seq with the Tuxedo Suite
 RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
Gene Expression analysis with RNA-Seq data
 Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
RNA-Seq Module 2 From QC to differential gene expression.
 RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNAseq and Variant discovery
 RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12
 Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Introduction to RNA-Seq. David Wood Winter School in Mathematics and Computational Biology July 1, 2013
 Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
ChIP-seq and RNA-seq
 ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
Applications of short-read
 Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
ChIP-seq and RNA-seq. Farhat Habib
 ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
RNAseq Differential Gene Expression Analysis Report
 RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
Sequencing applications. Today's outline. Hands-on exercises. Applications of short-read sequencing: RNA-Seq and ChIP-Seq
 Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Introduction to RNAseq Analysis. Milena Kraus Apr 18, 2016
 Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
 Background Wikipedia Lee and Mahadavan, JCB, 2009 History (Platform Comparison) P Park, Nature Review Genetics, 2009 P Park, Nature Reviews Genetics, 2009 Rozowsky et al., Nature Biotechnology, 2009
Background Wikipedia Lee and Mahadavan, JCB, 2009 History (Platform Comparison) P Park, Nature Review Genetics, 2009 P Park, Nature Reviews Genetics, 2009 Rozowsky et al., Nature Biotechnology, 2009
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
SCALABLE, REPRODUCIBLE RNA-Seq
 SCALABLE, REPRODUCIBLE RNA-Seq SCALABLE, REPRODUCIBLE RNA-Seq Advances in the RNA sequencing workflow, from sample preparation through data analysis, are enabling deeper and more accurate exploration
SCALABLE, REPRODUCIBLE RNA-Seq SCALABLE, REPRODUCIBLE RNA-Seq Advances in the RNA sequencing workflow, from sample preparation through data analysis, are enabling deeper and more accurate exploration
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ),
,") Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
RNA-Seq Analysis. Simon Andrews, Laura v
 RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
Bioinformatics Monthly Workshop Series. Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory
 Bioinformatics Monthly Workshop Series Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory Schedule for Fall, 2015 PILM Bioinformatics Web Server (09/21/2015)
Bioinformatics Monthly Workshop Series Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory Schedule for Fall, 2015 PILM Bioinformatics Web Server (09/21/2015)
RNAseq Applications in Genome Studies. Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford
 RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
Quantifying gene expression
 Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Long and short/small RNA-seq data analysis
 Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
RNA-sequencing. Next Generation sequencing analysis Anne-Mette Bjerregaard. Center for biological sequence analysis (CBS)
") RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
Introduction of RNA-Seq Analysis
 Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Canadian Bioinforma3cs Workshops
 Canadian Bioinforma3cs Workshops www.bioinforma3cs.ca Module #: Title of Module 2 1 Module 3 Expression and Differen3al Expression (lecture) Obi Griffith & Malachi Griffith www.obigriffith.org ogriffit@genome.wustl.edu
Canadian Bioinforma3cs Workshops www.bioinforma3cs.ca Module #: Title of Module 2 1 Module 3 Expression and Differen3al Expression (lecture) Obi Griffith & Malachi Griffith www.obigriffith.org ogriffit@genome.wustl.edu
RNA-Seq Workshop AChemS Sunil K Sukumaran Monell Chemical Senses Center Philadelphia
 RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
Cufflinks Assembly & DE v2.0 BaseSpace App Guide
 Cufflinks Assembly & DE v2.0 BaseSpace App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Workflow 5 Workflow Diagram for Global Analysis 6 Workflow Diagram for Differential
Cufflinks Assembly & DE v2.0 BaseSpace App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Workflow 5 Workflow Diagram for Global Analysis 6 Workflow Diagram for Differential
Analysis of RNA-seq Data. Feb 8, 2017 Peikai CHEN (PHD)
") Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Statistical Genomics and Bioinformatics Workshop. Genetic Association and RNA-Seq Studies
 Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Sequence Analysis 2RNA-Seq
 Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
!!!! The Genome Access Course. RNA-Seq Data Analysis!!!!!!!!!!!!
 RNA-Seq Data Analysis 3/26/15' Transcrip0onal'Profiling'Using' High'Throughput'Sequencing' Ben'King' Mount'Desert'Island'Biological'Laboratory' cdna'made' from'rna' RNA'Sequencing'(RNAKSeq)' Sequence'
RNA-Seq Data Analysis 3/26/15' Transcrip0onal'Profiling'Using' High'Throughput'Sequencing' Ben'King' Mount'Desert'Island'Biological'Laboratory' cdna'made' from'rna' RNA'Sequencing'(RNAKSeq)' Sequence'
Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data
 Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data Jan Hellemans 7th international qpcr & NGS Event - Freising March 24 th, 2015 Therapeutics lncrna oncology
Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data Jan Hellemans 7th international qpcr & NGS Event - Freising March 24 th, 2015 Therapeutics lncrna oncology
Eucalyptus gene assembly
 Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
NGS Data Analysis and Galaxy
 NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
Introduction to RNA-Seq in GeneSpring NGS Software
 Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Next Generation Sequencing
 Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
RNA Ribonucleic Acid. Week 14, Lecture 28. RNA- seq is a new, emerging field. Two major domains applica:on 12/4/ When the transcriptome is known
 2014 - BMMB 852D: Applied Bioinforma:cs RNA Ribonucleic Acid Week 14, Lecture 28 István Albert Biochemistry and Molecular Biology and Bioinforma:cs Consul:ng Center Penn State Two major domains applica:on
2014 - BMMB 852D: Applied Bioinforma:cs RNA Ribonucleic Acid Week 14, Lecture 28 István Albert Biochemistry and Molecular Biology and Bioinforma:cs Consul:ng Center Penn State Two major domains applica:on
Analysis of RNA-seq Data. Bernard Pereira
 Analysis of RNA-seq Data Bernard Pereira The many faces of RNA-seq Applications Discovery Find new transcripts Find transcript boundaries Find splice junctions Comparison Given samples from different experimental
Analysis of RNA-seq Data Bernard Pereira The many faces of RNA-seq Applications Discovery Find new transcripts Find transcript boundaries Find splice junctions Comparison Given samples from different experimental
De novo assembly in RNA-seq analysis.
 De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
Mapping Next Generation Sequence Reads. Bingbing Yuan Dec. 2, 2010
 Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
How to deal with your RNA-seq data?
 How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
Demo of mrna NGS Concluding Report
 Demo of mrna NGS Concluding Report Project: Demo Report Customer: Dr. Demo Company/Institute: Exiqon AS Date: 09-Mar-2015 Performed by Exiqon A/S Company Reg.No.(CVR) 18 98 44 31 Skelstedet 16 DK-2950,
Demo of mrna NGS Concluding Report Project: Demo Report Customer: Dr. Demo Company/Institute: Exiqon AS Date: 09-Mar-2015 Performed by Exiqon A/S Company Reg.No.(CVR) 18 98 44 31 Skelstedet 16 DK-2950,
GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment
 GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment Zhaojun Zhang, Shunping Huang, Jack Wang, Xiang Zhang, Fernando Pardo
GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment Zhaojun Zhang, Shunping Huang, Jack Wang, Xiang Zhang, Fernando Pardo
Introduction to transcriptome analysis using High Throughput Sequencing technologies. D. Puthier 2012
 Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Thesis/Dissertation Collections
 Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 5-19-2017 Identification of Differential Expression of p53 associated RNAs at 3 Different Treatment Timepoints,
Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 5-19-2017 Identification of Differential Expression of p53 associated RNAs at 3 Different Treatment Timepoints,
Transcriptional analysis of b-cells post flu vaccination using rna sequencing
 Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 3-20-2013 Transcriptional analysis of b-cells post flu vaccination using rna sequencing Manimozhi Manivannan Follow
Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 3-20-2013 Transcriptional analysis of b-cells post flu vaccination using rna sequencing Manimozhi Manivannan Follow
1. Introduction Gene regulation Genomics and genome analyses
 1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
Wheat CAP Gene Expression with RNA-Seq
 Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
Combined final report: genome and transcriptome assemblies
 Combined final report: genome and transcriptome assemblies Nadia Fernandez- Trinity assembly, RSEM, Tophat and Cufflinks/Cuffmerge/Cuffdiff pipeline, and MAKER annotation Stephanie Gutierrez Avril Harder
Combined final report: genome and transcriptome assemblies Nadia Fernandez- Trinity assembly, RSEM, Tophat and Cufflinks/Cuffmerge/Cuffdiff pipeline, and MAKER annotation Stephanie Gutierrez Avril Harder
NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript
 NIH Public Access Author Manuscript Published in final edited form as: Nat Protoc. ; 7(3): 562 578. doi:10.1038/nprot.2012.016. Differential gene and transcript expression analysis of RNA-seq experiments
NIH Public Access Author Manuscript Published in final edited form as: Nat Protoc. ; 7(3): 562 578. doi:10.1038/nprot.2012.016. Differential gene and transcript expression analysis of RNA-seq experiments
Analysis of RNA-seq Data
 Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Novel methods for RNA and DNA- Seq analysis using SMART Technology. Andrew Farmer, D. Phil. Vice President, R&D Clontech Laboratories, Inc.
 Novel methods for RNA and DNA- Seq analysis using SMART Technology Andrew Farmer, D. Phil. Vice President, R&D Clontech Laboratories, Inc. Agenda Enabling Single Cell RNA-Seq using SMART Technology SMART
Novel methods for RNA and DNA- Seq analysis using SMART Technology Andrew Farmer, D. Phil. Vice President, R&D Clontech Laboratories, Inc. Agenda Enabling Single Cell RNA-Seq using SMART Technology SMART
Introduction to transcriptome analysis using High Throughput Sequencing technologies. D. Puthier 2012
 Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 Transcriptome: the old school Cyanine 5 (Cy5) Cy-3: - Excitation 550nm - Emission 570nm Cy-5: - Excitation
Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 Transcriptome: the old school Cyanine 5 (Cy5) Cy-3: - Excitation 550nm - Emission 570nm Cy-5: - Excitation
Transcriptome Assembly, Functional Annotation (and a few other related thoughts)
") Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
The first thing you will see is the opening page. SeqMonk scans your copy and make sure everything is in order, indicated by the green check marks.
 Open Seqmonk Launch SeqMonk The first thing you will see is the opening page. SeqMonk scans your copy and make sure everything is in order, indicated by the green check marks. SeqMonk Analysis Page 1 Create
Open Seqmonk Launch SeqMonk The first thing you will see is the opening page. SeqMonk scans your copy and make sure everything is in order, indicated by the green check marks. SeqMonk Analysis Page 1 Create
RNA-Seq Analysis. August Strand Genomics, Inc All rights reserved.
 RNA-Seq Analysis August 2014 Strand Genomics, Inc. 2014. All rights reserved. Contents Introduction... 3 Sample import... 3 Quantification... 4 Novel exon... 5 Differential expression... 12 Differential
RNA-Seq Analysis August 2014 Strand Genomics, Inc. 2014. All rights reserved. Contents Introduction... 3 Sample import... 3 Quantification... 4 Novel exon... 5 Differential expression... 12 Differential
02 Agenda Item 03 Agenda Item
 01 Agenda Item 02 Agenda Item 03 Agenda Item SOLiD 3 System: Applications Overview April 12th, 2010 Jennifer Stover Field Application Specialist - SOLiD Applications Workflow for SOLiD Application Application
01 Agenda Item 02 Agenda Item 03 Agenda Item SOLiD 3 System: Applications Overview April 12th, 2010 Jennifer Stover Field Application Specialist - SOLiD Applications Workflow for SOLiD Application Application
Reference genomes and common file formats
 Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Reference genomes and common file formats
 Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
RNA-Seq de novo assembly training
 RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
Analytics Behind Genomic Testing
 A Quick Guide to the Analytics Behind Genomic Testing Elaine Gee, PhD Director, Bioinformatics ARUP Laboratories 1 Learning Objectives Catalogue various types of bioinformatics analyses that support clinical
A Quick Guide to the Analytics Behind Genomic Testing Elaine Gee, PhD Director, Bioinformatics ARUP Laboratories 1 Learning Objectives Catalogue various types of bioinformatics analyses that support clinical
RNA-Seq Tutorial 1. Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016
 RNA-Seq Tutorial 1 Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016 Slides available at www.msi.umn.edu/tutorial-materials RNA-Seq Tutorials Lectures RNA-Seq experiment
RNA-Seq Tutorial 1 Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016 Slides available at www.msi.umn.edu/tutorial-materials RNA-Seq Tutorials Lectures RNA-Seq experiment
Whole Transcriptome Analysis of Illumina RNA- Seq Data. Ryan Peters Field Application Specialist
 Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
SCIENCE CHINA Life Sciences. Comparative analysis of de novo transcriptome assembly
 SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 156 162 RESEARCH PAPER doi: 10.1007/s11427-013-4444-x Comparative analysis of de novo transcriptome assembly CLARKE Kaitlin 1, YANG
SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 156 162 RESEARCH PAPER doi: 10.1007/s11427-013-4444-x Comparative analysis of de novo transcriptome assembly CLARKE Kaitlin 1, YANG
Fully Automated Genome Annotation with Deep RNA Sequencing
 Fully Automated Genome Annotation with Deep RNA Sequencing Gunnar Rätsch Friedrich Miescher Laboratory of the Max Planck Society Tübingen, Germany Friedrich Miescher Laboratory of the Max Planck Society
Fully Automated Genome Annotation with Deep RNA Sequencing Gunnar Rätsch Friedrich Miescher Laboratory of the Max Planck Society Tübingen, Germany Friedrich Miescher Laboratory of the Max Planck Society
Introduction to NGS analyses
 Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Post-assembly Data Analysis
 Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
Workflows and Pipelines for NGS analysis: Lessons from proteomics
 Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
measuring gene expression December 5, 2017
 measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
SCIENCE CHINA Life Sciences
 SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 143 155 RESEARCH PAPER doi: 10.1007/s11427-013-4442-z Comparative study of de novo assembly and genome-guided assembly strategies for
SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 143 155 RESEARCH PAPER doi: 10.1007/s11427-013-4442-z Comparative study of de novo assembly and genome-guided assembly strategies for
RNA-Seq analysis workshop
 RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
Transcriptomics analysis with RNA seq: an overview Frederik Coppens
 Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
CSE 549: RNA-Seq aided gene finding
 CSE 549: RNA-Seq aided gene finding Finding Genes We ll break gene finding methods into 3 main categories. ab initio latin from the beginning w/o experimental evidence comparative make use of knowledge
CSE 549: RNA-Seq aided gene finding Finding Genes We ll break gene finding methods into 3 main categories. ab initio latin from the beginning w/o experimental evidence comparative make use of knowledge
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS
 SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Software for Estimation of Human Transcriptome Isoform Expression Using RNA-Seq Data
 University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses Spring 5-18-2012 Software for Estimation of Human Transcriptome Isoform Expression
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses Spring 5-18-2012 Software for Estimation of Human Transcriptome Isoform Expression
BST 226 Statistical Methods for Bioinformatics David M. Rocke. March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
Basics of RNA-Seq. (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility
 Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility") 2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
TECH NOTE Stranded NGS libraries from FFPE samples
 TECH NOTE Stranded NGS libraries from FFPE samples Robust performance with extremely degraded FFPE RNA (DV 200 >25%) Consistent library quality across a range of input amounts (5 ng 50 ng) Compatibility
TECH NOTE Stranded NGS libraries from FFPE samples Robust performance with extremely degraded FFPE RNA (DV 200 >25%) Consistent library quality across a range of input amounts (5 ng 50 ng) Compatibility
RNA standards v May
 Standards, Guidelines and Best Practices for RNA-Seq: 2010/2011 I. Introduction: Sequence based assays of transcriptomes (RNA-seq) are in wide use because of their favorable properties for quantification,
Standards, Guidelines and Best Practices for RNA-Seq: 2010/2011 I. Introduction: Sequence based assays of transcriptomes (RNA-seq) are in wide use because of their favorable properties for quantification,
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. GEP goals: Evidence Based Annotation. Evidence for Gene Models 12/26/2018
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Introduction to Next Generation Sequencing
 The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
RNA-seq data analysis with Chipster. Eija Korpelainen CSC IT Center for Science, Finland
 RNA-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? How to operate the Chipster software Short introduction to RNA-seq Analyzing RNA-seq
RNA-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? How to operate the Chipster software Short introduction to RNA-seq Analyzing RNA-seq
RNA-Seq data analysis course September 7-9, 2015
 RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
measuring gene expression December 11, 2018
 measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and
measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and