L3: Short Read Alignment to a Reference Genome
|
|
|
- Meghan Phillips
- 6 years ago
- Views:
Transcription


1 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017

2 Where to get help! Read the posting guide before sending !
3 Overview Understand the difference between reference genome builds Introduction to Illumina sequencing Short read aligners BWA Bowtie STAR Other aligners Coverage and Depth Mappability Use of decoy and sponge databases Alignment Quality, SAMStat, Qualimap Samtools and Picard tools, Visualization of alignment data A very brief look at long reads, graph genome aligners and de novo genome assembly
4 Reference Genomes A haploid representation of a species genome. The human genome is a haploid mosaic derived from 13 volunteer donors from Buffalo, NY. USA. In regions where there is known large scale population variation, sets of alternate loci (178 in GRCh38) are assembled alongside the reference locus. The current build has around 500 gaps, whereas the first version had ~150,000 gaps. Genome Reference Consortium:

5

6 600 30X

7 Sequencers
8 Illumina sequencers

9 Illumina Genome Analyzer
10 Illumina sequencing technology Illumina sequencing is based on the Solexa technology developed by Shankar Balasubramanian and David Klenerman (1998) at the University of Cambridge. Multiple steps in Sequencing by synthesis (explained in next slide) Library Preparation Bridge amplification and Cluster generation Sequencing using reversible terminators Image acquisition and Fastq generation Alignment and data analysis


11 Illumina Flowcell

12 Sequencing By Synthesis technology


13 Illumina Sequencing Bridge Amplification Sequencing Cluster growth Incorporation of fluorescence, reversibly terminated tagged nt
14 Multiplexing Multiplexing gives the ability to sequence multiple samples at the same time. Useful when sequencing small genomes or specific genomic regions. Different barcode adaptors are ligated to different samples. Reads de-multiplexd after sequencing.

 for the sequence in Line 2. Historically there are a number of different FASTQ formats.")
15 FASTQ format A FASTQ file normally uses four lines per sequence. Line-1 begins with a '@' character and is followed by a sequence identifier and an optional description. Line-2 is the raw sequence letters. Line-3 begins with a '+' character and is optionally followed by the same sequence identifier again. Line-4 encodes the quality scores (ASCII) for the sequence in Line 2. Historically there are a number of different FASTQ formats. These include the Sanger Format, Illumina/Solexa 1.0, Illumina 1.3, 1.5, 1.8 and 1.9 Cock et al., Nucleic Acids Res Apr;38(6):
16 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudoaligners for RNA-seq quantification Kallisto Salmon Sailfish
17 More than 90 Short Read Aligners
18 Short Read Aligners
19 Download and install BWA # download wget # extract tar -xvfj bwa a.tar.bz2 # x extracts, v is verbose (details of what it is doing), f skips prompting for each individual file, and j tells it to unzip.bz2 files cd bwa a Make # Add BWA to your PATH by editing ~/.bashrc file (or.bash_profile or.profile file) export PATH=$PATH:/path/to/bwa a source ~/.bashrc # /path/to/ is a placeholder. Replace with real path to BWA on your machine # manual man./bwa.1
20 BWA Burrows-Wheeler transform (BWT) algorithm with FM-index using suffix arrays. BWA can map low-divergent sequences against a large reference genome, such as the human genome. It consists of three algorithms: BWA-backtrack (Illumina sequence reads up to 100bp) BWA-SW (more sensitive when alignment gaps are frequent) BWA-MEM (maximum exact matches) BWA SW and MEM can map longer sequences (70bp to 1Mbp) and share similar features such as long-read support and split alignment, but BWA-MEM, which is the latest, is generally recommended for high-quality queries as it is faster and more accurate. BWA-MEM also has better performance than BWA-backtrack for bp Illumina reads. Li and Durbin, 2009, Bioinformatics Need to prepare a genome index
21 Bowtie2 Bowtie2 handles reads longer than 50 nt. Given a reference and a set of reads, this method reports at least one good local alignment for each read if one exists. Indexing: Since genomes and sequencing datasets are usually large, dynamic programing proves to be inefficient and high-memory machines are required, with lots of secondary storage, etc. Uses Burrows-Wheeler Transform (BWT) The transform is performed by sorting all rotations of the test and these acts as the index for the sequence. The aim is to find out from which part of the genome a the read originates. Need to prepare a genome index.
22

23 STAR: Splicing Transcripts Alignment to a Reference Non-contiguous nature of transcripts, presence of splice-forms make short read (36-200nt) RNA-seq alignment to a genome challenging. Reads contain mismatches, insertions and deletions caused by genomic variation and sequencing errors. Mapping spliced sequence from non contiguous genomic regions. Multi-mapping reads Two steps: Seed searching and clustering/stitching/scoring (find MMP -maximal mappable prefix using Suffix Arrays) Fast splice aware aligner, high memory (RAM) footprint Can detect chimeric transcripts Generate indices using a reference genome fasta, and annotation gtf or gff from Ensembl/UCSC genome browsers Dobin et al., 2013 Bioinformatics
24 Normalised Counts Do not use RPKM (Reads Per Kilobase Million) and FPKM (Fragments Per Kilobase Million) to express normalised counts in ChIP-seq (or RNA-seq). CPM (Counts Per Million) and TPM (Transcripts Per MIllion) is the less biased way of normalising read counts. When calculating TPM, the only difference from RPKM is that you normalize for gene/transcript length first, and then normalize for sequencing depth second. However, the effects of this difference are quite profound. RPKM vs TPM Lior Pachtor video
25 Coverage and Depth Coverage: average number of reads of a given length that align to or cover known reference bases with the assumption that the reads are randomly distributed across the genome. Depth: redundancy of coverage or the total number of bases sequenced and aligned at a given reference position. Increased depth of coverage rescues inadequacies of sequencing methods. Sims et al., 2014, Nat. Rev. Genet.

26 Lander-Waterman model of Coverage Coverage = Read Length * Number of reads / haploid Genome length
27 Mappability *Calculated based on 30nt sequence tags Rozowsky, (2009) Not all of the genome is available for mapping when reads are aligned to the unmasked genome. Alignability: This provide a measure of how often the sequence found at the particular location will align within the whole genome. Uniqueness: This is a direct measure of sequence uniqueness throughout the reference genome.
28 Decoy and Sponge databases The decoy contains human sequences missing from the hg19 reference, mitochondrial sequences and viral sequences integrated into the human genome. blog article on decoys The sponge contains ribosomal and mitochondrial sequences, non-centromeric Huref sequences absent in GRCh38 (hg38), centromeric models etc (Miga et al., 2015). These mop up ambiguous sequences, resulting in more accurate and faster alignment.
29 Processing SAM / BAM files SAMtools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format. import: SAM-to-BAM conversion view: BAM-to-SAM conversion and sub alignment retrieval sort: sorting alignment merge: merging multiple sorted alignments index: indexing sorted alignment faidx: FASTA indexing and subsequence retrieval tview: text alignment viewer pileup: generating position-based output and consensus/indel calling RSamTools package in Bioconductor allows similar functionality in R.
30 Picard tools Picard is a collection of Java-based command-line utilities that manipulate sequencing data and formats such as SAM/BAM/CRAM and VCF. It has a Java API (SAM-JDK) for creating new programs that read and write SAM files. The mark duplicate function is particularly useful. Picard tools

31 SAMStat for mapping QC SAMstat is a C program that plots nucleotide overrepresentation and other statistics in mapped and unmapped reads and helps understand the relationship between potential protocol biases and poor mapping. It reports statistics for unmapped, poorly and accurately mapped reads separately. This allows for identification of a variety of problems, such as remaining linker and adaptor sequences, causing poor mapping Lassmann et al., 2011, Bioinformatics.
 ChIP-seq http://qualimap.bioinfo.")
32 Qualimap Qualimap provides both a GUI and a command-line interface to facilitate the quality control of alignment sequencing data and feature counts. Supported types of experiments include: Whole-genome sequencing Whole-exome sequencing RNA-seq (special mode available) ChIP-seq

33 Visualizing binding sites and replicates Integrated Genome Viewer (IGV)
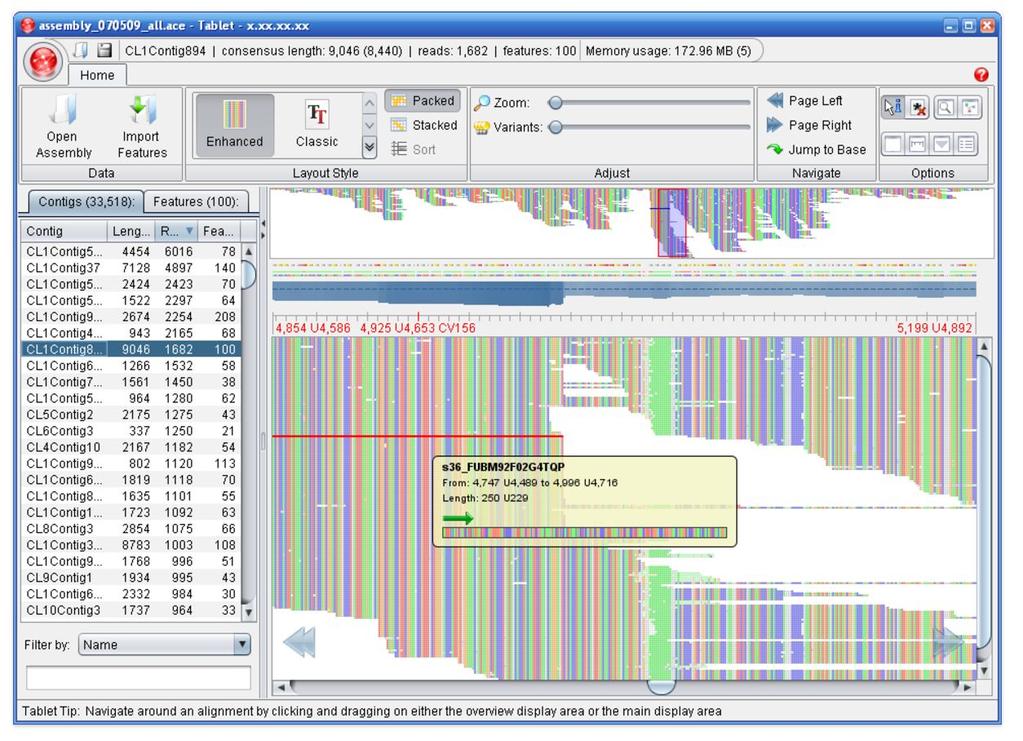
34 Visualization: Tablet

35 How to get external sequencing data via SRA toolkit Extract data sets from the Sequence Read Archive or dbgap (NCBI) These repositories store sequencing data in the SRA format Prefetch: fetch fastq data Fastq-dump: Convert SRA data into fastq format sam-dump: Convert SRA data to SAM format sra-stat: Generate statistics about SRA data (quality distribution, etc.) vdb-validate: Validate the integrity of downloaded SRA data
 De novo assembly of genomes (usually using De Bruijn graph methods for species without")
36 The Future Graph based reference genomes and aligners are beginning to make an appearance and will eventually replace linear genome representations. Long read sequencing technologies (Oxford Nonopore, Pacific Bioscience, Illumina and others) De novo assembly of genomes (usually using De Bruijn graph methods for species without reference genomes) is an alternative to mapping.
Short Read Alignment to a Reference Genome
 Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Transcriptomics analysis with RNA seq: an overview Frederik Coppens
 Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Quantifying gene expression
 Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Lecture 7. Next-generation sequencing technologies
 Lecture 7 Next-generation sequencing technologies Next-generation sequencing technologies General principles of short-read NGS Construct a library of fragments Generate clonal template populations Massively
Lecture 7 Next-generation sequencing technologies Next-generation sequencing technologies General principles of short-read NGS Construct a library of fragments Generate clonal template populations Massively
C3BI. VARIANTS CALLING November Pierre Lechat Stéphane Descorps-Declère
 C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, March 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 1: Introduction into high-throughput
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, March 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 1: Introduction into high-throughput
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
Mapping Next Generation Sequence Reads. Bingbing Yuan Dec. 2, 2010
 Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Introduction of RNA-Seq Analysis
 Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Introduction to transcriptome analysis using High Throughput Sequencing technologies. D. Puthier 2012
 Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
NEXT GENERATION SEQUENCING. Farhat Habib
 NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
Next Generation Sequencing. Tobias Österlund
 Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
RNA-Seq Analysis. Simon Andrews, Laura v
 RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
Introduction to RNA-Seq. David Wood Winter School in Mathematics and Computational Biology July 1, 2013
 Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Sequence Analysis 2RNA-Seq
 Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
BST 226 Statistical Methods for Bioinformatics David M. Rocke. March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
TECH NOTE Ligation-Free ChIP-Seq Library Preparation
 TECH NOTE Ligation-Free ChIP-Seq Library Preparation The DNA SMART ChIP-Seq Kit Ligation-free template switching technology: Minimize sample handling in a single-tube workflow >> Simplified protocol with
TECH NOTE Ligation-Free ChIP-Seq Library Preparation The DNA SMART ChIP-Seq Kit Ligation-free template switching technology: Minimize sample handling in a single-tube workflow >> Simplified protocol with
Alignment methods. Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics
 Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
RNA-sequencing. Next Generation sequencing analysis Anne-Mette Bjerregaard. Center for biological sequence analysis (CBS)
") RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
How to deal with your RNA-seq data?
 How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
Next Generation Sequencing: An Overview
 Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
ChIP-seq and RNA-seq. Farhat Habib
 ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
Applications of short-read
 Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
ChIP-seq and RNA-seq
 ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
Why QC? Next-Generation Sequencing: Quality Control. Illumina data format. Fastq format:
 Why QC? Next-Generation Sequencing: Quality Control BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute Do you want to include the reads with low quality base calls?
Why QC? Next-Generation Sequencing: Quality Control BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute Do you want to include the reads with low quality base calls?
RNAseq Differential Gene Expression Analysis Report
 RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNA-Seq Workshop AChemS Sunil K Sukumaran Monell Chemical Senses Center Philadelphia
 RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
Introduc)on to Bioinforma)cs of next- genera)on sequencing. Sequence acquisi)on and processing; genome mapping and alignment manipula)on
on to Bioinforma)cs of next- genera)on sequencing. Sequence acquisi)on and processing; genome mapping and alignment manipula)on") Introduc)on to Bioinforma)cs of next- genera)on sequencing Sequence acquisi)on and processing; genome mapping and alignment manipula)on Ruslan Sadreyev Director of Bioinformatics Department of Molecular
Introduc)on to Bioinforma)cs of next- genera)on sequencing Sequence acquisi)on and processing; genome mapping and alignment manipula)on Ruslan Sadreyev Director of Bioinformatics Department of Molecular
Next-Generation Sequencing: Quality Control
 Next-Generation Sequencing: Quality Control Bingbing Yuan BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Why QC? Do you want to
Next-Generation Sequencing: Quality Control Bingbing Yuan BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Why QC? Do you want to
NGS in Pathology Webinar
 NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
Genomic DNA ASSEMBLY BY REMAPPING. Course overview
 ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
Sequencing applications. Today's outline. Hands-on exercises. Applications of short-read sequencing: RNA-Seq and ChIP-Seq
 Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
SMRT Analysis Barcoding Overview (v6.0.0)
") SMRT Analysis Barcoding Overview (v6.0.0) Introduction This document applies to PacBio RS II and Sequel Systems using SMRT Link v6.0.0. Note: For information on earlier versions of SMRT Link, see the document
SMRT Analysis Barcoding Overview (v6.0.0) Introduction This document applies to PacBio RS II and Sequel Systems using SMRT Link v6.0.0. Note: For information on earlier versions of SMRT Link, see the document
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Data Basics. Josef K Vogt Slides by: Simon Rasmussen Next Generation Sequencing Analysis
 Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Measuring transcriptomes with RNA-Seq
 Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2017 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC
Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2017 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC
Gene Expression analysis with RNA-Seq data
 Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
DATA FORMATS AND QUALITY CONTROL
 HTS Summer School 12-16th September 2016 DATA FORMATS AND QUALITY CONTROL Romina Petersen, University of Cambridge (rp520@medschl.cam.ac.uk) Luigi Grassi, University of Cambridge (lg490@medschl.cam.ac.uk)
HTS Summer School 12-16th September 2016 DATA FORMATS AND QUALITY CONTROL Romina Petersen, University of Cambridge (rp520@medschl.cam.ac.uk) Luigi Grassi, University of Cambridge (lg490@medschl.cam.ac.uk)
ChIP-seq analysis 2/28/2018
 ChIP-seq analysis 2/28/2018 Acknowledgements Much of the content of this lecture is from: Furey (2012) ChIP-seq and beyond Park (2009) ChIP-seq advantages + challenges Landt et al. (2012) ChIP-seq guidelines
ChIP-seq analysis 2/28/2018 Acknowledgements Much of the content of this lecture is from: Furey (2012) ChIP-seq and beyond Park (2009) ChIP-seq advantages + challenges Landt et al. (2012) ChIP-seq guidelines
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
RNAseq Applications in Genome Studies. Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford
 RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
Reference genomes and common file formats
 Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
High-Throughput Bioinformatics: Re-sequencing and de novo assembly. Elena Czeizler
 High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Total RNA isola-on End Repair of double- stranded cdna
 Total RNA isola-on End Repair of double- stranded cdna mrna Isola8on using Oligo(dT) Magne8c Beads AAAAAAA A Adenyla8on (A- Tailing) A AAAAAAAAAAAA TTTTTTTTT AAAAAAA TTTTTTTTT TTTTTTTT TTTTTTTTT AAAAAAAA
Total RNA isola-on End Repair of double- stranded cdna mrna Isola8on using Oligo(dT) Magne8c Beads AAAAAAA A Adenyla8on (A- Tailing) A AAAAAAAAAAAA TTTTTTTTT AAAAAAA TTTTTTTTT TTTTTTTT TTTTTTTTT AAAAAAAA
Wheat CAP Gene Expression with RNA-Seq
 Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
Variation detection based on second generation sequencing data. Xin LIU Department of Science and Technology, BGI
 Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer. Project XX1001. Customer Detail
 Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer Project XX Customer Detail Table of Contents. Bioinformatics analysis pipeline...3.. Read quality check. 3.2. Read alignment...3.3.
Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer Project XX Customer Detail Table of Contents. Bioinformatics analysis pipeline...3.. Read quality check. 3.2. Read alignment...3.3.
Basics of RNA-Seq. (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility
 Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility") 2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
Introduction to NGS analyses
 Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
ChIP-seq data analysis with Chipster. Eija Korpelainen CSC IT Center for Science, Finland
 ChIP-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? Short introduction to ChIP-seq Analyzing ChIP-seq data Central concepts Analysis
ChIP-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? Short introduction to ChIP-seq Analyzing ChIP-seq data Central concepts Analysis
RNA-Seq Module 2 From QC to differential gene expression.
 RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
High performance sequencing and gene expression quantification
 High performance sequencing and gene expression quantification Ana Conesa Genomics of Gene Expression Lab Centro de Investigaciones Príncipe Felipe Valencia aconesa@cipf.es Next Generation Sequencing NGS
High performance sequencing and gene expression quantification Ana Conesa Genomics of Gene Expression Lab Centro de Investigaciones Príncipe Felipe Valencia aconesa@cipf.es Next Generation Sequencing NGS
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Genome 373: Mapping Short Sequence Reads II. Doug Fowler
 Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 1: Introduction into high-throughput
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 1: Introduction into high-throughput
1. Introduction Gene regulation Genomics and genome analyses
 1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
Next-generation sequencing technologies
 Next-generation sequencing technologies NGS applications Illumina sequencing workflow Overview Sequencing by ligation Short-read NGS Sequencing by synthesis Illumina NGS Single-molecule approach Long-read
Next-generation sequencing technologies NGS applications Illumina sequencing workflow Overview Sequencing by ligation Short-read NGS Sequencing by synthesis Illumina NGS Single-molecule approach Long-read
Next Generation Sequencing Lecture Saarbrücken, 19. March Sequencing Platforms
 Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA
 TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA Stranded, Illumina ready library construction in
TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA Stranded, Illumina ready library construction in
Next Generation Sequencing
 Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
Illumina (Solexa) Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme
 Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme") Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
BST227 Introduction to Statistical Genetics. Lecture 8: Variant calling from high-throughput sequencing data
 BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
RNA-Seq de novo assembly training
 RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
Alignment. J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
 Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Short Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 Introduction to Short Read Alignment UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Short Read Alignment UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased
 has increased") Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
Incorporating Molecular ID Technology. Accel-NGS 2S MID Indexing Kits
 Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
BICF Variant Analysis Tools. Using the BioHPC Workflow Launching Tool Astrocyte
 BICF Variant Analysis Tools Using the BioHPC Workflow Launching Tool Astrocyte Prioritization of Variants SNP INDEL SV Astrocyte BioHPC Workflow Platform Allows groups to give easy-access to their analysis
BICF Variant Analysis Tools Using the BioHPC Workflow Launching Tool Astrocyte Prioritization of Variants SNP INDEL SV Astrocyte BioHPC Workflow Platform Allows groups to give easy-access to their analysis
Bulked Segregant Analysis For Fine Mapping Of Genes. Cheng Zou, Qi Sun Bioinformatics Facility Cornell University
 Bulked Segregant Analysis For Fine Mapping Of enes heng Zou, Qi Sun Bioinformatics Facility ornell University Outline What is BSA? Keys for a successful BSA study Pipeline of BSA extended reading ompare
Bulked Segregant Analysis For Fine Mapping Of enes heng Zou, Qi Sun Bioinformatics Facility ornell University Outline What is BSA? Keys for a successful BSA study Pipeline of BSA extended reading ompare
Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA. March 2, Steven R. Kain, Ph.D. ABRF 2013
 Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA March 2, 2013 Steven R. Kain, Ph.D. ABRF 2013 NuGEN s Core Technologies Selective Sequence Priming Nucleic Acid Amplification
Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA March 2, 2013 Steven R. Kain, Ph.D. ABRF 2013 NuGEN s Core Technologies Selective Sequence Priming Nucleic Acid Amplification
CM581A2: NEXT GENERATION SEQUENCING PLATFORMS AND LIBRARY GENERATION
 CM581A2: NEXT GENERATION SEQUENCING PLATFORMS AND LIBRARY GENERATION Fall 2015 Instructors: Coordinator: Carol Wilusz, Associate Professor MIP, CMB Instructor: Dan Sloan, Assistant Professor, Biology,
CM581A2: NEXT GENERATION SEQUENCING PLATFORMS AND LIBRARY GENERATION Fall 2015 Instructors: Coordinator: Carol Wilusz, Associate Professor MIP, CMB Instructor: Dan Sloan, Assistant Professor, Biology,
RNA Sequencing: Experimental Planning and Data Analysis. Nadia Atallah September 12, 2018
 RNA Sequencing: Experimental Planning and Data Analysis Nadia Atallah September 12, 2018 A Next Generation Sequencing (NGS) Refresher Became commercially available in 2005 Construction of a sequencing
RNA Sequencing: Experimental Planning and Data Analysis Nadia Atallah September 12, 2018 A Next Generation Sequencing (NGS) Refresher Became commercially available in 2005 Construction of a sequencing
Introduction to metagenome assembly. Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014
 Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12
 Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
 Background Wikipedia Lee and Mahadavan, JCB, 2009 History (Platform Comparison) P Park, Nature Review Genetics, 2009 P Park, Nature Reviews Genetics, 2009 Rozowsky et al., Nature Biotechnology, 2009
Background Wikipedia Lee and Mahadavan, JCB, 2009 History (Platform Comparison) P Park, Nature Review Genetics, 2009 P Park, Nature Reviews Genetics, 2009 Rozowsky et al., Nature Biotechnology, 2009
NGS, Cancer and Bioinformatics. 5/3/2015 Yannick Boursin
 NGS, Cancer and Bioinformatics 5/3/2015 Yannick Boursin 1 NGS and Clinical Oncology NGS in hereditary cancer genome testing BRCA1/2 (breast/ovary cancer) XPC (melanoma) ERCC1 (colorectal cancer) NGS for
NGS, Cancer and Bioinformatics 5/3/2015 Yannick Boursin 1 NGS and Clinical Oncology NGS in hereditary cancer genome testing BRCA1/2 (breast/ovary cancer) XPC (melanoma) ERCC1 (colorectal cancer) NGS for
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
solid S Y S T E M s e q u e n c i n g See the Difference Discover the Quality Genome
 solid S Y S T E M s e q u e n c i n g See the Difference Discover the Quality Genome See the Difference With a commitment to your peace of mind, Life Technologies provides a portfolio of robust and scalable
solid S Y S T E M s e q u e n c i n g See the Difference Discover the Quality Genome See the Difference With a commitment to your peace of mind, Life Technologies provides a portfolio of robust and scalable
Measuring transcriptomes with RNA-Seq. BMI/CS 776 Spring 2016 Anthony Gitter
 Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu Overview RNA-Seq technology The RNA-Seq quantification problem Generative
Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu Overview RNA-Seq technology The RNA-Seq quantification problem Generative
NGS part 2: applications. Tobias Österlund
 NGS part 2: applications Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
NGS part 2: applications Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Introduction to Next Generation Sequencing
 The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
Alignment & Variant Discovery. J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014
 Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Analysis Datasheet Exosome RNA-seq Analysis
 Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
NUCLEIC ACIDS. DNA (Deoxyribonucleic Acid) and RNA (Ribonucleic Acid): information storage molecules made up of nucleotides.
 and RNA (Ribonucleic Acid): information storage molecules made up of nucleotides.") NUCLEIC ACIDS DNA (Deoxyribonucleic Acid) and RNA (Ribonucleic Acid): information storage molecules made up of nucleotides. Base Adenine Guanine Cytosine Uracil Thymine Abbreviation A G C U T DNA RNA 2
NUCLEIC ACIDS DNA (Deoxyribonucleic Acid) and RNA (Ribonucleic Acid): information storage molecules made up of nucleotides. Base Adenine Guanine Cytosine Uracil Thymine Abbreviation A G C U T DNA RNA 2
CNV and variant detection for human genome resequencing data - for biomedical researchers (II)
") CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
Aaron Liston, Oregon State University Botany 2012 Intro to Next Generation Sequencing Workshop
 Output (bp) Aaron Liston, Oregon State University Growth in Next-Gen Sequencing Capacity 3.5E+11 2002 2004 2006 2008 2010 3.0E+11 2.5E+11 2.0E+11 1.5E+11 1.0E+11 Adapted from Mardis, 2011, Nature 5.0E+10
Output (bp) Aaron Liston, Oregon State University Growth in Next-Gen Sequencing Capacity 3.5E+11 2002 2004 2006 2008 2010 3.0E+11 2.5E+11 2.0E+11 1.5E+11 1.0E+11 Adapted from Mardis, 2011, Nature 5.0E+10
Introduction to human genomics and genome informatics
 Introduction to human genomics and genome informatics Session 1 Prince of Wales Clinical School Dr Jason Wong ARC Future Fellow Head, Bioinformatics & Integrative Genomics Adult Cancer Program, Lowy Cancer
Introduction to human genomics and genome informatics Session 1 Prince of Wales Clinical School Dr Jason Wong ARC Future Fellow Head, Bioinformatics & Integrative Genomics Adult Cancer Program, Lowy Cancer