RNA-Seq data analysis
|
|
|
- Jeffry Goodman
- 6 years ago
- Views:
Transcription






1 RNA-Seq data analysis Material Slides: pdf : one per page pdf : three per page with comment lines Memento: Hands on: Data files: Results files: The speakers Sarah Maman Christine Gaspin Céline Noirot Matthias Zytnicki Claire Hoede Cédric Cabau Nathalie Villa-Vialaneix (pour la partie biostat)
2 Session organisation Day 1 Morning (9h00-12h30) : - Prerequisite unix/format Exercises Day 2 Morning (9h00-12h30) : - Spliced read mapping Exercises and Visualisation - Biological reminds - Expression quantification Theory + exercises Afternoon (14h-17h) : - Sequence quality Theory + exercises - Spliced read mapping Theory Afternoon (14h-17h) : - mrna calling Theory + exercises - Models comparison Theory + exercises Prerequisite unix Summary - Unix reminders Genotoul infrastructure organisation How to connect to genotoul How to transfer data From the web to genotoul From genotoul to your computer How to launch jobs on the cluster...

 (100 MB user quota) /work/ HPC computational disk space (TEMPORARY) (1 TB user quota) /save/ User disk space (with BACKUP) (250 GB user")
3 Vocabulary : Cluster / Node Cluster : set of nodes Node : Huge computer (with several CPUs) CPU CPU CPU CPU «genotoul» cluster node001 à 068 : 2720 INTEL cores, 17TB of memory Ceri001 à 034 : 1632 AMD cores 12TB of memory qlogin qrsh ssh Bigmem01 : 64 INTEL cores 1TB of memory smp : 240 INTEL cores 3TB of memory Internet «genotoul» login nodes computer nodes Storage Facilities Genotoul Bioinfo Disk spaces /usr/local/bioinfo/src Bioinformatics Software /bank/ International genomics Databanks /home/ User configuration files (ONLY) (100 MB user quota) /work/ HPC computational disk space (TEMPORARY) (1 TB user quota) /save/ User disk space (with BACKUP) (250 GB user quota)
 Usage of software installed Free access to computational cluster Own space disk (/save & /work")
4 How to connect to genotoul? Xming (Windows graphic) + Putty (Connection) MobaXterm (Executable file) command line: in a Terminal windows ssh username@genotoul.toulouse.inra.fr Linux Linux account Access to a work environment Login + password Share resources (Cpu, memory, disk) Usage of software installed Free access to computational cluster Own space disk (/save & /work directory) Linux Which are the main unix/linux commands you know?
5 Very Important Tips Copy / Paste with the mouse Select a text (it is automatically copied) Click on the mouse wheel (the text is pasted where the cursor is located) Command and path completion : Use the TAB key Back to the previously used commands : Use the «up» and «down» keys OGE (Open Grid Engine) Queues availables for users Queue Priority Max time Max slots workq (default) everyone Access H 4120 unlimitq everyone 100 unlimited 680 smpq on demand 0 unlimited 240 hypermemq on demand 0 unlimited 96 Interq (qlogin) everyone H 40 galaxyq galaxy users No node shared unlimited 120 OGE (Open Grid Engine) Characteristics of work working space Workq 1 core 8 GB memory maximum Write only /work directory (temporary disk space) Work space 1 TB quota disk per user (on /work directory) 120 days files without access automatic purged Time resource constraint H annually computing time (more on demand)
6 OGE (Open Grid Engine) qlogin (with display) / qrsh or qrsh -X Connected Disconnected [laborie@genotoul2 ~]$ qlogin Your job ("QLOGIN") has been submitted waiting for interactive job to be scheduled... Your interactive job has been successfully scheduled. Establishing /SGE/ogs/inra/tools/qlogin_wrapper.sh session to host node [laborie@node001 ~]$ [laborie@node001 ~]$ exit logout /SGE/ogs/inra/tools/qlogin_wrapper.sh exited with exit code 0 [laborie@genotoul2 ~]$ OGE (Open Grid Engine) Job Submission : some examples Default (workq, 1 core, 8 GB memory max) $qsub myscript.sh Your job ("mon_script.sh") has been submitted More memory (workq, 1 core, 32 / 36 GB memory) $qsub -l mem=32g -l h_vmem=36g myscript.sh Your job ("mon_script.sh") has been submitted More cores (workq, 8 core, 8*8 GB memory) $qsub -l parallel smp 8 myscript.sh Your job ("mon_script.sh") has been submitted OGE (Open Grid Engine) Job Submission : some examples Script edition $nedit myscript.sh ### head of myscript.sh ### #!/bin/bash #$ -m a #$ -l mem=32g #$ -l h_vmem=36g #Mon programme commence ici ls ### end of myscript.sh ### Submission $qsub myscript.sh Your job ("mon_script.sh") has been submitted
7 OGE (Open Grid Engine) qsub : batch Submission 1 - First write a script (ex: myscript.sh) with the command line as following: #$ -N job_name to give a name to the job #$ -o /work/.../output_file_name to redirect output standard #$ -e /work/.../error_file_name error_file_name : to redirect error file #$ -q workq queue_name : to specify the batch queue #$ -m bea mail sending : (b:begin, a:abort, e:end) #$ -l mem=8g to ask for 8GB of mem (minimum reservation) #$ -l h_vmem=10g to fix the maximum consumption of memory # My command lines I want to run on the cluster blastall -d swissprot -p blastx -i /save/.../z72882.fa 2 - Then submit the job with the qsub command line as following: $qsub myscript.sh Your job ("mon_script.sh") has been submitted OGE (Open Grid Engine) Monitoring jobs : qstat $qstat job-id prior name user state submit/start queue slots ja-task-id Job-ID : prior : name : user : state : submit/start at : Queue : slots : ja-task-id : job identifier priority of job job name user name actual state of job (see follow) submit/start date batch queue name number of slots aked for the job job array task identifier (see follow) OGE (Open Grid Engine) Monitoring jobs : qstat state : actually state of job d(eletion) : job is deleting E(rror) : job is in error state h(old), w(waiting) : job is pending t(ransferring) : job is about to be executed r(unning) : job is running man qstat : to see all options of qstat command
8 OGE (Open Grid Engine) Deleting a job : qdel $qstat -u laborie job-id prior name user state submit/start at queue slots ja-task-id sleep laborie r 02/25/ :23:03 workq@node002 1 $ qdel laborie has registered the job for deletion Array of jobs concept Concept : segment a job into smaller atomic jobs Improve the processing time very significantly (the calculation is performed on multiple processing cores) blast in job array mode split... blastx+ -d nt -i seq1.fa blastx+ -d nt -i seq2.fa blastx+ -d nt -i seq3.fa for i in... qarray script.sh Execution on 3 cores

9 Downloading / transferring Several possible cases Internet browser ( + save as) Winscp / scp wget «genotoul» server Downloading / transferring File download from Internet to «genotoul server»: Copy the URL of the file to download wget Downloading / transferring Transfer between genotoul and desktop computer We recommend to use «scp» command (secure copy) scp [user@host1:]file1 [user@host2:]file2 copy file from the network Example copy from desktop to "genotoul": scp source_name bleuet@genotoul:destination_name

10 Downloading / transferring WinSCP / FileZilla : copy via graphical interface Introduction to NGS formats Summary - Format remind

11 fastq format The standard for storing outputs of HTS A text-based format for storing a read and its corresponding quality scores. 1 read <-> 4 1:N:0:ATTCAGAATAATCTTA NCTAAGTGTTAGGGGGTTTCCGCCCCTTAGTGCTGCAGCTAACGCATTAAGCACTCCGCCTGGGGAGTACGGTCGCAAGACTGAAAA + #<3?BFGGGGGGEGGGGGGGEGGGGGG@F1FGGGGGGDDGG1FB</9FE=EGGGGGGGG>GGGGBGGGGG<<C/BDGGGGGGC=GGG Begins with '@' character and is followed by a sequence identifier The raw sequence Begins with a '+' character and is optionally followed by the same sequence identifier Encodes the quality values for th read, contains the same number of symbols as letters in the read fastq format la proba d'une erreur : Sequence Alignment/Map (SAM) format Data sharing was a major issue with the 1000 genomes Capture all of the critical information about NGS data in a single indexed and compressed file Generic alignment format Supports short and long reads (454 Solexa Solid) Flexible in style, compact in size, efficient in random access Website : Paper : Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, [PMID: ]


12 Sequence Alignment/Map (SAM) format 11 mandatory fields Variable number of optional fields Fields are tab delimited Sequence Alignment/Map (SAM) format SAM format - Flag field Decimal values in sam lines Picard tools
13 How to manipulate them? Samtools Picard tools Bedtools Hands-on : unix & formats Training accounts : anemone aster camelia chardon cobee cosmos dahlia geranium arome bleuet capucine clematite coquelicot cyclamen digitale gerbera Exercice 1 : using basic unix commands Exercice 2 : format manipulation Summary - Biological reminders Context, vocabulary, transcriptome variability... Methods to analyse transcriptomes What is RNAseq? High throughput sequencers Illumina protocol, paired-end library, directional library Retrieve public data and presentation of data for practical work

14 Different approaches : Alignment to De novo Reference transcriptome No reference genome, no transcriptome available Very expensive computationally Lots of variation in results depending on the software used Most are incomplete Computationally inexpensive Reference genome When available Allow reads to align to unannotated sites Computationally expensive Need a spliced aligner Context Prerequis : Reference genome available RNAseq sequencing (sequence of transcript) Try to answer to : How to map transcript to the genome? How to discover new transcript? What are the alternative transcript? Source : en.wikipedia.org/wiki/user:forluvoft/sandbox Vocabulary Gene : functional units of DNA that contain the instructions for generating a functional product. UTR Exon1 Promoter region Intron Exon2 Intron UTR Exon3 TSS Exon : coding region of mrna included in the transcript Intron : non coding region TSS : Transcription Start Site 1st amino acid Transcript : stretch of DNA transcribed into an RNA molecule

15 Transcription products Protein coding gene: transcribed in mrna ncrna : highly abundant and functionally important RNA trna, rrna, snornas, micrornas, sirnas, pirnas lincrna ENCODE Alternative splicing Alternative splicing (or differential splicing) the exons are reconnected in multiple ways during RNA splicing. different mrnas translated into different protein isoforms a single gene may code for multiple proteins. Intron Retention Post-transcriptional modification (eukaryotic cells) eg: the conversion of precursor messenger RNA into mature mrna (mrna), editing...

16 Transcript degradation mrna export to the cytoplasm, protected from degradation by a 5 cap structure and a 3 polya tail. the polya tail is gradually shortened by exonucleases the degradation machinery rapidly degrades the mrna in both in directions. others mechanisms, bypass the need for deadenylation and can remove the mrna from the transcriptional pool independently. Cis-natural antisense transcript Natural antisense transcripts (NATs) are a group of RNAs encoded within a cell that have transcript complementarity to other RNA transcripts. Fusion genes A fusion gene is a hybrid gene formed from two previously separate genes. It can occur as the result of a translocation, interstitial deletion, or chromosomal inversion. Often, fusion genes are oncogenes. They often come from trans-splicing : Trans-splicing is a special form of RNA processing in eukaryotes where exons from two different primary RNA transcripts are joined end to end and ligated.
17 Transcriptome variability Many types of transcripts (mrna, ncrna...) Many isoform (non canonical splice sites, intron retention...) Number of transcripts 6 possible variation factor between transcripts: 10 or more, expression variation between samples. Allele specific expression How can we study the transcriptome? Techniques classification PCR/RT-QPCR EST SAGE MicroArrays No quantification Indirect quantification Low throughput Low throughput (up to hundreds) Low throughput (up to thousands) High throughput (up to millions) Discovery (Yes) No No Discovery (Yes) Need transcript sequence partially known Difficulties in discovering novels splice events What is different with RNA-Seq? No prior knowledge of sequence needed Specificity of what is measured Increased dynamic range of measure, more sensitive detection Direct quantification Good reproducibility Different levels : genes, transcripts, allele specificity, structure variations New feature discovery: transcripts, isoforms, ncrna, structures (fusion...) Possible detection of SNPs,...
 Discover isoform")
18 SGS platforms SGS platforms PacBio : ISOseq Produce full-length transcripts without assembly (up to 10 kb in length) Discover isoform Can not be used for differential expression analysis


19 MinION Available now for sequencing cdnas Longest read length: 98kb Median read length: 1kb Mean read length: 2kb Coming next: direct analysis of RNA RNA modifications PCR-free protocols Increased accuracy compared to using reverse transcriptases What are we looking for? Identify genes List new genes Identify transcripts List new alternative splice forms Quantify these elements differential expression Usual questions on RNA-Seq! How many replicates? How many reads for each sample? How many conditions for a full transcriptome? How long should my reads be? Single-end or paired-end? Technical or/and biological replicates?
 : Experiments should be performed with two or more biological replicates, unless there is a compelling reason why this is impractical or wasteful Replicate concordance: the gene")

20 Depth VS Replicates Encode (2016) : Experiments should be performed with two or more biological replicates, unless there is a compelling reason why this is impractical or wasteful Replicate concordance: the gene level quantification should have a Spearman correlation of >0.9 between isogenic (same donor) replicates and >0.8 between anisogenic (different donor) replicates. Between 30M and 100M reads per sample depending on the study. evaluate the similarity between the transcriptional profiles of two polya+ samples ==> modest depths of sequencing. discovery of novel transcribed elements and strong quantification of known transcript isoforms ==> more extensive sequencing. Zhang et al : From 3 replicates improve DE detection and control false positive rate. Depth VS Replicates Illumina RNA-Seq protocol 1 Flowcell: in general 1 run equivalent to 8 Lane Hiseq 2500: 2 Billion reads single or 4 Billion paired reads.



21 RNA-Seq library preparation Clusters generation / Sequencing Paired-end sequencing Modification of the standard single-read DNA library preparation facilitates reading both ends of each fragment Improvement of mapping Help to detect structural variations in the genome like insertions or deletions, copy number variations, and genome rearrangements
. The fragment are usually forward-reverse. Strand specific RNA-Seq protocol Retrieve public data Why?")
22 Paired-end VS single-end Single-end Paired-end sequence fragment 1 sequence fragment 1 (forward) (c)dna sequence fragment 2 (rev) alignement genome gene The cdna size hive the insert size (ex pb). The fragment are usually forward-reverse. Strand specific RNA-Seq protocol Retrieve public data Why? Because there s a lot of public data that would be sufficient for your analysis The authors often use only part of the data to answer their own problems Perhaps you don t need your own data


23 Retrieve public data Retrieve public data Retrieve public data

24 Retrieve public data Retrieve public data Retrieve public data
")

25 Retrieve public data Retrieve public data prefetch <sra_accession> --max-size (20G by default) fastq-dump sra_file.sra Summary - Sequence quality Known RNAseq biais How to check the quality? How to clean the data?

 Hexamer random priming bias Hexamer random priming bias A strong distinctive pattern in the")

26 RNAseq specific bias Influence of the library preparation Random hexamer priming Positional bias and sequence specificity bias. Robert et al. Genome Biology, 2011,12:R22 Transcript length bias Some reads map to multiple locations (??) Hexamer random priming bias Hexamer random priming bias A strong distinctive pattern in the nucleotide frequencies of the first 13 positions at the 5'-end : sequence specificity of the polymerase due to the end repair performed Reads beginning with a hexamer over-represented in the hexamer distribution at the beginning relative to the end are down-weighted



27 Hexamer random effect Orange = reads start sites Blue = coverage Transcript length bias the differential expression of longer transcripts is more likely to be identified than that of shorter transcripts Bias mappability Quality of the reference genome influence results assembly finishing Sequence composition Repeated sequences Annotation quality




.")




28 Verifying RNA-Seq quality FastQC : Has been developed for genomic data fastqqc Report The analysis in FastQC is performed by a series of analysis modules. Quick evaluation of whether the results of the module seem : entirely normal (green tick), slightly abnormal (orange triangle) or very unusual (red cross). These evaluations must be taken in the context of what you expect from your library. A 'normal' sample as far as FastQC is concerned is random and diverse. fastqqc Report Statistics per Base Sequence Quality This view shows an overview of the range of quality values across all bases at each position in the FastQ file. Common to see base calls falling into the orange area towards the end of a read.







29 fastqqc Report Statistics per Sequence Quality Score See if a subset of your sequences have universally low quality values. fastqqc Report Statistics per Base N Content This module plots out the percentage of base calls at each position for which an N was called. Usual to see a very low proportion of Ns appearing nearer the end of a sequence. fastqqc Report Statistics Per Base Sequence Content Per Base Sequence Content plots out the proportion of each base position in a file for which each of the four normal DNA bases has been called. In a random library : little/no difference between the different bases of a sequence run, so the lines in this plot should run parallel with each other. If strong biases which change : overrepresented sequence contaminating your library. RNA-seq OK






30 fastqqc Report Statistics per Base GC Distribution Per Base GC Content plots out the GC content of each base position in a file. Random library : little/no difference between the different bases of a sequence run => plot horizontally. The overall GC content should reflect the GC content of the underlying genome. GC bias: changes in different bases, overrepresented sequence contaminating your library. => plot not horizontally. RNA-seq OK fastqqc Report Statistics per Sequence GC Content This module measures the GC content across the whole length of each sequence in a file and compares it to a modeled normal distribution of GC content. fastqqc Report Statistics per Sequence Length Distribution Some sequence fragments contain reads of wildly varying lengths. Even within uniform length libraries some pipelines will trim sequences to remove poor quality base calls from the end.


 Adaptor presence Alignment on reference for the")
, Length of the reads expected (150pb),")
31 fastqqc Report Statistics per Duplicate Sequences High level of duplication indicate an enrichment biais. RNA-seq OK fastqqc Report Overrepresented Kmers A kmer is a subsequence of length k Should spot overrepresented sequences, give a good impression of any contamination. Kmers showing a rise towards the end of the library indicate progressive contamination with adapters. Check for adaptor sequence or poly-a sequence RNA-seq OK Take home message on quality analysis Elements to be checked : Random priming effect K-mer (polya, polyt) Adaptor presence Alignment on reference for the second quality check and filtering. A good run?: Expected number of reads produced (2x 2 billions / flowcell), Length of the reads expected (150pb), Random selection of the nucleotides and the GC%, Good alignment: very few unmapped reads, pairs mapped on opposite strands.
. cutadapt -a ADAPTER [options] [-o output.fastq] input.fastq Input file : fasta, fastq or compressed (gz, bz2, xz).")
32 Cleaning analysis Cleaning : Low quality bases Adaptors Software : Trim_galore Cutadapt Trimmomatic Sickle PRINSEQ... Cutadapt Searches & removes adapter & tag in all reads. Trim quality Filter too short or untrimmed reads (in a separate output file). cutadapt -a ADAPTER [options] [-o output.fastq] input.fastq Input file : fasta, fastq or compressed (gz, bz2, xz). Cutadapt Cutadapt supports trimming of paired-end reads, trimming both reads in a pair at the same time. Processing both files at the same time is highly recommended.
33 Hands-on: quality control Data for the exercises: - from Mohammed Zouine (ENSAT) - tomato wild type and mutant type (without seeds) with the transcription factor Sl-ARF8 (auxine response factor 8) overexpressed - clonal lineage - paired, 100 pb non stranded - triplicated - in the publication process - subsampled on chromosome 6 for faster analysis Use FastQC and cutadapt Exercice 3 : quality control of used datasets Exercice 4: cleaning used datasets Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Mapping Novel Tr/Gn quantif. Sample 1 Novel Tr/Gn quantif. Sample N Mapping Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Novel Tr/Gn quantif. Sample 1 Mapping Novel Tr/Gn quantif. Sample N
34 Summary Spliced read mapping & Visualisation What is a spliced aligner? Reference genome & transcriptome files formats Tophat principle principle and usage BAM & Bed files formats Visualisation with IGV Aim Spliced read mapping & Visualisation Aim: Discover the true location (origin) of each read on the reference. Problems: Some features (repetitive regions, assembly errors, missing information) make it impossible for some reads. Reads may be split by potentially thousands of bases of intronic sequence. And: Do it in/with reasonable time/resources. Splice sites Canonical splice site: which accounts for more than 99% of splicing GT and AG for donor and acceptor sites Non-canonical site: GC-AG splice site pairs, AT-AC pairs Trans-splicing: splicing that joins two exons that are not within the same RNA transcript



 for reads and for junctions")
35 Hard case Lot of variations (sequencing errors, mutations) Repeats Reads spanning 3+ exons Gene or pseudogene Small end anchor Kim et al, Genome Biology, 2013 Unknown junction inside poorly rarely expressed gene Most used tools Tools for splice-mapping: - Tophat: - : Comparing tools How to compare tools? sensibility (maximize #mapped reads) specificity (assign reads to the correct position) for reads and for junctions processing time memory requirement All of these are conflicting criteria...


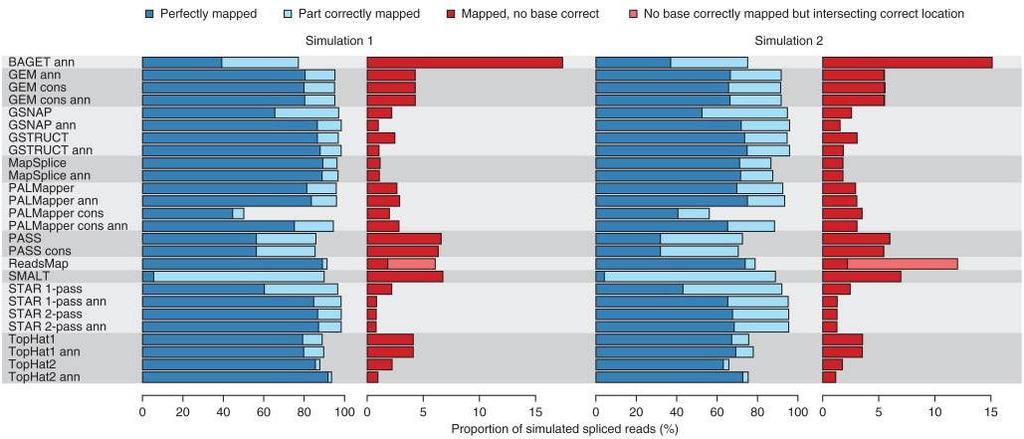
36 RGASP3 The RNA-seq Genome Annotation Assessment Project Engström et al., Nature Methods, 2013 RGASP3 The RNA-seq Genome Annotation Assessment Project Engström et al., Nature Methods, 2013 RGASP3 The RNA-seq Genome Annotation Assessment Project Engström et al., Nature Methods, 2013
 Mapping reads using index FA index")

37 Other benchmark Basically similar conclusions... Mapping steps Indexing reference (only once) Mapping reads using index FA index FQ BD map BAM TopHat pipeline Numerous steps to resolve hard cases Each step uses of heuristics with parameters users have to define a value u/software/topha t Kim et al, Genome Biology, 2013



38 An other aligner : Spliced Transcripts Alignment to a Reference Outperforms other aligners by more than a factor of 50 in mapping speed Another strategy: search for a MMP from the 1st base MMP search repeated for the unmapped portion next to the junction do it in both fwd and rev directions cluster seeds from the mates of paired-end RNA-seq reads Soft-clipping is the main difference between Tophat and Dobin et al, Bioinformatics, 2011 Two passes strategy «Improved ability to align reads by short spanning lengths is sufficient to explain the quantification benefit of two-pass alignment» Veeneman et al, Bioinformatics, 2016
39 indexing Hands-on: Type and count the number of options. Core command: --runmode genomegenerate --genomedir genome_dir --genomefastafiles genome.fasta To use N CPUs, add: --runthreadn N If you have an annotation: --sjdbgtffile annot.gtf Some precomputed indices are already available: bin//genomes or on your preferred platform: /bank/db Where to find a reference genome? Retrieving the genome file (fasta): The Genome Reference Consortium NCBI chromosome naming with not well supported by mapping software Prefer EMBL Reference transcriptome file What is a GTF file? An annotation file: loci of coding genes (transcripts, CDS, UTRs), non-coding genes, etc. Gene Transfer Format (derived from GFF): chr source ENSEMBL ENSEMBL ENSEMBL ENSEMBL feature exon exon exon exon start end score.... strand frame.... [attributes] gene_id "ENSG01"; gene_id "ENSG01"; gene_id "ENSG01"; gene_id "ENSG02"; transcript_id transcript_id transcript_id transcript_id ENST01.1 ; ENST01.1 ; ENST01.2 ; ENST02.1 ; gene_name gene_name gene_name gene_name "ABC"; "ABC"; "ABC"; "DEF"; ENST01.1 ENSG02, ENST02.1, DEF ENST01.2 ENSG01, ABC - gene_id value : unique identifier for the gene. - transcript_id value : unique identifier for the transcript. The chromosome names should be the same in the gtf file and fasta files (e.g. chr1 vs Chr1 vs 1).
40 Hands-on : Exercice n 2 A/ Create a directory for the genome and annotation files. Get the FASTA and GTF files from: seq/data/reference/ Create the index. Tip: you can allocate N CPUs with the qsub/qrsh option -pe parallel_smp N mapping Core command: --genomedir genome_dir --readfilesin reads1.fastq reads2.fastq [--sjdbgtffile annot.gtf --runthreadn n] If the read files are gzipped (reads1.fq.gz): --readfilescommand zcat Intron options: genomic gap is considered intron if --alignintronmin [21] --alignintronmax [500000] Max. number of mismatches: --outfiltermismatchnmax [10] Default options are probably tuned for mammalian genomes. SAM / BAM formats Sequence Alignment/Map format: Each line stores an alignment/map Coor ref +r001/1 +r002 +r003 +r004 -r003 -r001/ AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT TTAGATAAAGGATA*CTG aaaagataa*ggata gcctaagctaa ATAGCT...TCAGC ttagcttaggc CAGCGGCAT name flag chr start mapq cigar nnext snext tlen seq qual tags r ref M2I4M1D3M = TTAGATAAAGGATACTG * r002 0 ref S6M1P1I4M * 0 0 AAAAGATAAGGATA * r003 0 ref S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0; r004 0 ref M14N5M * 0 0 ATAGCTTCAGC * r ref H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1; r ref M = 7-39 CAGCGGCAT * NM:i:1 Header stores genome VN:1.5 SN:ref LN:45
41 Fields Coor ref AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT +r001/1 +r002 +r003 +r004 -r003 -r001/2 TTAGATAAAGGATA*CTG aaaagataa*ggata gcctaagctaa ATAGCT...TCAGC ttagcttaggc CAGCGGCAT name flag chr start mapq cigar nnext snext tlen seq qual tags r ref M2I4M1D3M = TTAGATAAAGGATACTG * r002 0 ref S6M1P1I4M * 0 0 AAAAGATAAGGATA * r003 0 ref S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0; r004 0 ref M14N5M * 0 0 ATAGCTTCAGC * r ref H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1; r ref M = 7-39 CAGCGGCAT * NM:i:1 Flags: MapQ: similar to a phred score nnext: = means same chr In general, * means NA CIGAR Coor ref +r001/1 +r002 +r003 +r004 -r003 -r001/ AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT TTAGATAAAGGATA*CTG aaaagataa*ggata gcctaagctaa ATAGCT...TCAGC ttagcttaggc CAGCGGCAT name flag chr start mapq cigar nnext snext tlen seq qual tags r ref M2I4M1D3M = TTAGATAAAGGATACTG * r002 0 ref S6M1P1I4M * 0 0 AAAAGATAAGGATA * r003 0 ref S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0; r004 0 ref M14N5M * 0 0 ATAGCTTCAGC * r ref H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1; r ref M = 7-39 CAGCGGCAT * NM:i:1 30M means 30 matches or mismatches I and D: insertion/deletion S and H: soft/hard clipping Tags Coor ref +r001/1 +r002 +r003 +r004 -r003 -r001/ AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT TTAGATAAAGGATA*CTG aaaagataa*ggata gcctaagctaa ATAGCT...TCAGC ttagcttaggc CAGCGGCAT name flag chr start mapq cigar nnext snext tlen seq qual tags r ref M2I4M1D3M = TTAGATAAAGGATACTG * r002 0 ref S6M1P1I4M * 0 0 AAAAGATAAGGATA * r003 0 ref S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0; r004 0 ref M14N5M * 0 0 ATAGCTTCAGC * r ref H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1; r ref M = 7-39 CAGCGGCAT * NM:i:1 Format: 2-letter name:format:value (many different) NM: # mismatches SA: chimeric reads NH, HI: # hits for this sequence, hit index AS: alignment score nm: # mismatches per fragment
42 SAM / BAM BAM (Binary Alignment/Map) format: Compressed binary representation of SAM Greatly reduces storage space requirements to about 27% of original SAM samtools: reading, writing, and manipulating BAM files Most tools require a sorted and indexed BAM file. output options Output format: --outsamtype BAM SortedByCoordinate [SAM] Add more tags: --outsamattributes All Default output file name: Aligned.bam Modify prefix: --outfilenameprefix prefix Infer strand using intron motifs (for Cufflinks) --outsamstrandfield intronmotif [None] Start IH at --outsamattrihstart 0 [1] (for Cufflinks) other options Remove reads that did not pass the junction filter: --outfiltertype BySJOut [Normal] Filter out alignments with non-canonical intron motifs --outfilterintronmotifs RemoveNoncanonical Output SAM/BAM alignments to transcriptome into a separate file (for RSEM) --quantmode TranscriptomeSAM Two passes mode: is run once and discover new junctions. is run again, knowing the new junctions. (Probably most useful for poorly annotated genomes.)
43 Outputs Outputs (w/o specific options except BAM SortedByCoordinate): Aligned.sortedByCoord.out.bam: list of read alignments in SAM format compressed Log.out: main log file with a lot of detailed information about the run (for troubleshooting) Log.progress.out: reports job progress statistics Log.final.out: summary mapping statistics after mapping job is complete, very useful for quality control. SJ.out.tab: contains high confidence collapsed splice junctions in tab-delimited format (chr, intron start, end, strand, intron motif, in database, # uniquely mapping reads, # multi, max. overhang) technical issues Temporary disk space: Indexing the mouse genome requires 128GB and 1 hour on 6 slots. Mapping a 16M paired-end reads requires 110GB and 4 mins on 6 slots. New platform cluster: 34 cluster nodes with 4 12 cores and 384 GB of ram per node: 1632 cores 1 hypermem node (32 cores and 1024 GB of ram) A scratch file system (157 To available, 6 Gbps bandwith) Hands-on : Exercice n 2 B/ Map the 2 FASTQ files. Do not forget to provide a different output file name for each set. Index the output BAM files with: samtools index file.bam Get some stats with: samtools flagstat file.bam

44 Quasi-mapping: Sailfish Patro et al, Nat. Biotech., 2014 Reads are not mapped. Transcriptome is cut into small chunks of small k-mers. Same for reads. Take a k-mer from a transcript, counts how many times you find it in reads. Average the counts over a transcript. Resolve ambiguous counts. Quasi-mapping: why? Bray et al, Nat. Biotech., 2016 Other (most used) tool: kallisto, salmon Quasi-mapping: limitations Heavily relies on a good annotation: Unannotated genes will not be counted and may bias other genes counts. Does not align reads: Cannot find variation (SNP) in the reads.
. You can fetch some others through the server.")
45 Visualizing alignments on IGV Step 1: set the genome Exercice n 2 C/ Open the Genomes menu Choose Load Genome from File Provide your FASTA file. Some updated fields: Genome Chromosome Locus Tips: Some chromosomes are bundled with IGV (but they should have the same chromosome names). You can fetch some others through the server. Step 2: add the tracks Open the File menu Choose Load from File Provide your GTF file. Provide your BAM files (the BAI file should be also present). Some interesting loci: Go to locus: SL2.40ch06:34,298,666-34,306,292 Thin lines indicate introns. Notice that gene introns match with read introns. Notice that the first and last exons seems longer than annotation. It s probably not annotated UTR.
46 Explore IGV Zoom in/out Go right/left Hover over the reads and get some info. Notice (colored) genome variations. Change panel height. Go to next TSS with Ctrl+F (Ctrl+B for previous TSS) Go to SL2.40ch06:34,209,900-34,260,000 Look at the strand of the gene. Expand the gene track. Do you think the annotation is complete here? Which condition is more expressed? Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Mapping Novel Tr/Gn quantif. Sample 1 Novel Tr/Gn quantif. Sample N Mapping Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Novel Tr/Gn quantif. Sample 1 Mapping Novel Tr/Gn quantif. Sample N
47 : estimation of expression based on a read count. BAM files Gene A Gene A Gene B 5 2 Table file Gene B GTF file Estimation of: gene expression transcript expression exon expression Difficult cases Multi-mapping read Every quantification tools uses its own rules! Raw counts vs estimation Raw count vs estimation: what to do with ambiguous reads? ambiguous specific Gene A Gene B Pros estimation: Use more reads. More accurate? Cons estimation: Underlying model inaccurate. Raw counts for differential expression does not matter much.



48 Transcript expression Trapnell C et al. Nature Biotechnology 2010; 28: Raw counts tool: featurecounts Levels : exon, transcript, gene Multiple option for : Paired reads Assignation of reads Oriented library Also exists: HTseq-Count Estimation tool: RSEM Exhaustive tool Levels : transcript, gene May be used without reference genome (RNA-Seq de novo) Also exists: cufflinks
49 RSEM workflow Two main steps: rsem-prepare-reference rsem-calculate-expression In default use case, RSEM maps the reads (with Bowtie). Using a different mapping tool () requires: Extra parameters for Extra parameters for RSEM Hands-in: prepare reference Exercice n 3 Command line: /usr/local/bioinfo/src/rsem/rsem-1.3.0/rsem-p repare-reference --gtf annot.gtf genome.fasta rsem_lib Output files: rsem_lib.grp, rsem_lib.ti, rsem_lib.seq, and rsem_lib.chrlist are for internal use. rsem_lib.idx.fa: the transcript sequences rsem_lib.n2g.idx.fa: same, with N G Hands-in: calculate expression Command line: /usr/local/bioinfo/src/rsem/rsem-1.3.0/rsem-c alculate-expression --alignments alignment.bam rsem_lib quant Outputs: quant.isoforms.results: isoform level expression estimates quant.genes.results: same for genes quant.stat: directory with stats on various aspects of this step
50 Hands-in: calculate expression Other parameters: --paired-end: specify paired-end reads -p N: use N CPUs --seed N: seed for random number generators --calc-ci: calculate 95% credibility intervals and posterior mean estimates. --ci-memory 30000: size in MB of the buffer used for computing CIs --estimate-rspd: estimate the read start position distribution --no-bam-output: do not output any BAM file (produced by internal mapper) Output file format effective_length: # positions that can generate a fragment expected_count: read count, with mapping prob. and read qual TPM: Transcripts Per Million, relative transcript abundance, see infra FPKM: Fragments Per Kilobase of transcript per Million mapped reads, see infra IsoPct: isoform percentage posterior_mean_count, posterior_standard_deviation_of_count, pme_tpm, pme_fpkm: estimates calculated Gibbs sampler Output file format IsoPct_from_pme_TPM: isoform percentage calculated from pme_tpm values TPM_ci_lower_bound, TPM_ci_upper_bound, FPKM_ci_lower_bound, FPKM_ci_upper_bound: bounds of 95% credibility intervals TPM_coefficient_of_quartile_variation, RPKM_coefficient_of_quartile_variation: coefficients of quartile variation, a robust way of measuring the ratio between the standard deviation and the mean
51 RPKM vs FPKM vs TPM RPKM: Reads Per Kb of transcript per Million mapped r = # reads on a gene k = size of the gene (in kb) m = # reads in the sample (in millions) RKPM = r / (k m) FPKM: Fragments Per Kilobase Same with f = # fragments (2 reads in PE) on a gene Meaning: If you sequence at depth 106, you will have x = FPKM fragments of a 1kb-gene. RPKM vs FPKM vs TPM TMP: ri = # reads on a gene i si = size of the gene i cpbi = ri / si cpb = cpbi TMPi = cpbi / cpb 106 Remark: TMPi = FPKMi / ( FPKMj) 106 Meaning: If you have 106 transcripts, x = TMPi will originate from gene i. RPKM vs FPKM vs TPM These are refinement of library size normalization, with gene length effect. RPKM should not be used for PE reads. TMP tend to be favored now w.r.t. R/FPKM. None of them should be used for differential expression: only raw counts. Ask your questions to the stats guys.

52 Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Mapping Novel Tr/Gn quantif. Sample 1 Novel Tr/Gn quantif. Sample N Mapping Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Novel Tr/Gn quantif. Sample 1 Novel Tr/Gn quantif. Sample N Mapping New transcriptome: why?



53 Transcript reconstruction Gene location Exon location Junctions : - between read pair junction - within read junction Model building strategies Cufflinks assembles transcripts estimates their abundances: based on how many reads support each one last version: cufflinks 2.2.1, released May 05, 2014
: nb incompatible read = mini nb of transcripts needed each path = set of mutually compatible fragments overlapping each other Trapnell")

54 Cufflinks transcript assembly Transcripts assembly: fragments are divided into non-overlapping loci each locus is assembled independently Cufflinks assembler find the mini nb of transcripts that explain the reads find a minimum path cover (Dilworth's theorem): nb incompatible read = mini nb of transcripts needed each path = set of mutually compatible fragments overlapping each other Trapnell C et al. Nature Biotechnology 2010 Cufflinks transcript assembly Transcripts assembly: identification of incompatible fragments originated from distinct isoforms connection of compatible fragments in an overlap graph assembling isoforms from the overlap graph: here minimally covered by three paths, each representing a different isoform Trapnell C et al. Nature Biotechnology 2010 Cufflinks transcript assembly Reference Annotation Based Transcripts Assembly Assembling novel transcripts in the context of an existing annotation Roberts et al. Bioinformatics 2011
![Cufflinks inputs and options Command line: cufflinks [options] <aligned_reads.(sam/bam)> Some options: -h/--help -o/--output-dir -p/--num-threads -G/--GTF <reference_annotation.](/docs-images/71/66169300/images/55-2.jpg "(gtf/gff)> estimate isoform expression, no novel transcripts -g/--gtf-guide <reference_annotation.")
55 Cufflinks inputs and options Command line: cufflinks [options] <aligned_reads.(sam/bam)> Some options: -h/--help -o/--output-dir -p/--num-threads -G/--GTF <reference_annotation.(gtf/gff)> estimate isoform expression, no novel transcripts -g/--gtf-guide <reference_annotation.(gtf/gff)> use reference transcript annotation to guide assembly --max-bundle-length [3,500,000] --max-bundle-frags [500,000] --library-type library prep used for input reads Cufflinks library types Cufflinks library types
 Cufflinks GTF description transcripts.")
56 Cufflinks outputs transcripts.gtf contains assembled isoforms (coordinates and abundances) genes.fpkm_tracking contains the genes FPKM isoforms.fpkm_tracking contains the isoforms FPKM skipped.gtf contains skipped loci (too many fragments) Cufflinks GTF description transcripts.gtf (coordinates and abundances): contains assembled isoforms can be visualized with a genome viewer attributes: ids, FPKM, confidence interval, read coverage & support score: most abundant isoform = 1000 minor isoforms = minor FPKM/major FPKM cov: estimate for depth across the transcript Cufflinks GTF description transcripts.gtf (coordinates and abundances): visualization in IGV
57 Cufflinks / Cuffcompare Compare assemblies between conditions: compare your assembled transcripts to a reference annotation track Cufflinks transcripts across multiple experiments Command: cuffcompare [-r <reference.gtf>] [-o <outprefix>] <input1.gtf> Outputs: overall summary statistics union of all transfrags <cuff_in>.refmap - transfrags matching to reference transcript <cuff_in>.tmap - best reference transcript for each transfrag <outprefix>.tracking - tracking transfrags across samples <outprefix>.stats - <outprefix>.combined.gtf - Cuffcompare Class code de cuffcompare = complete match c contained j novel isoform e single exon i within intron o exonic overlap p polymerase run-on r repeat u unknown, intergenic x exonic overlap on the opposite strand s intronic overlap on the opposite strand > 50% lower case bases Cufflinks / Cuffmerge Merge together several assemblies: merge novel isoforms and known isoforms filters a number of transfrags that are probably artifacts build a new gene model describing all conditions Command: cuffmerge [options] -o <assembly_gtf_list> Options: -o/--output-dir -g/--ref-gtf -s/--ref-sequence --min-isoform-fraction discard isoforms with abundance below this [0.05] -p/--num-threads
:")





")
58 Cufflinks / Cuffmerge merged.gtf (coordinates and legacy): contains merged input assemblies can be visualized with a genome viewer attributes: ids, name, oid, nearest_ref, class_code, tss_id, p_id Tuxedo protocol StringTie assembles transcripts StringTie identified 36-60% more transcripts than the next best assembler (Cufflinks) last version: stringtie 1.3.3, released Feb 15, 2017

![bam> [options] Some options: -o [<path/>]<out.gtf> -G <ref_ann.](/docs-images/71/66169300/images/59-2.jpg "gff> --rf --fr - stranded library fr-firststrand fr-secondstrand -p")


59 StringTie transcript assembly Pertea et al. Nature Biotechnology 2015 StringTie Command: stringtie <aligned_reads.bam> [options] Some options: -o [<path/>]<out.gtf> -G <ref_ann.gff> --rf --fr - stranded library fr-firststrand fr-secondstrand -p <int> --merge - transcript merge mode Main output: GTF file containing the assembled transcripts Gene abundances in tab-delimited format Fully covered transcripts matching the reference annotation Files required as input to Ballgown In merge mode, a merged GTF file from a set of GTF files StringTie protocol
60 StringTie / gffcompare Command: gffcompare [-r <reference.gtf>] [-o <outprefix>] <input1.gtf> Some options: -R for -r option consider only the reference transcripts that overlap any of the input transfrags (Sn correction) -Q for -r option consider only the input transcripts that overlap any of the reference transcripts (Precision correction); discard all "novel" loci Output: cuffcompare like output files StringTie / gffcompare strtcmp.stats (transcript assembly accuracy comparison) #= Summary for dataset: stringtie_asm.gtf # Query mrnas: in loci (17231 multi-exon transcripts) # (3731 multi-transcript loci, ~1.3 transcripts per locus) # Reference mrnas : in loci (15850 multi-exon) # Super-loci w/ reference transcripts: # Sensitivity Precision Base level: Exon level: Intron level: Intron chain level: Transcript level: Locus level: gffcompare2highcharts.pl Command: gffcompare2highcharts.pl --stats STATS_FILE[...,STATS_FILE_n] > output.html
61 Hands-on: transcripts assembly Using cufflinks et al: Exercise 7: reconstruct known and novel transcripts Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Mapping Novel Tr/Gn quantif. Sample 1 Novel Tr/Gn quantif. Sample N Mapping Analysis workflow Sample 1 RNA-seq reads Transcript modelling cufflinks Mapping Tr/Gn RSEM Merge cuffmerge Tr/Gn RSEM Sample N RNA-seq reads Tr/Gn RSEM Ref Tr/Gn quantif. Sample 1 Ref. gene Genome annotation Mapping Mapping Ref Tr/Gn quantif. Sample N Novel gene Genome annotation Tr/Gn RSEM Transcript modelling cufflinks Novel Tr/Gn quantif. Sample 1 Mapping Novel Tr/Gn quantif. Sample N
62 Hands-on : star, RSEM with new gtf Exercice n 8 (Optional) Commands : Star and RSEM: see exercice n 5 and 6 How to choose count matrix? Quality of the annotation : do not forget to check the genes structure with IGV presence of genes of interest too many transcripts quality metrics with gffcompare Number of reads mapped Number of reads assigned Jflow Workflow management system Launch a workflow with one command line Available on the Genotoul platform /usr/local/bioinfo/src/jflow/jflow/bin/jflow_cli.py <workflow_name> <workflow_parameters...>
 --reference-genome fasta-file=reference.fasta (index-directory=/path/to/directory) --gtf-file model.")
 --modeling-software [cufflinks stringtie] (default: cufflinks) Rna-Seq Workflows on Jflow Launch workflow:")
 --gtf-file model.")
63 Rna-Seq Workflows on Jflow Rna-Seq Workflows on Jflow Launch workflow: /usr/local/bioinfo/src/jflow/jflow/bin/jflow_cli.p y rnaseq --sample reads-1=myfile_r1.fastq.gz (reads-2=myfile_r2.fastq.gz) --reference-genome fasta-file=reference.fasta (index-directory=/path/to/directory) --gtf-file model.gtf --protocol (illumina_stranded, other) default : illumina_stranded Others parameters: --trim-reads : to trim reads before proceeding default: TruSeq Adapter --compute-gtf-model : to compute a new gtf model (gtf-file parameter is optional in this case) --modeling-software [cufflinks stringtie] (default: cufflinks) Rna-Seq Workflows on Jflow Launch workflow: /usr/local/bioinfo/src/jflow/jflow/bin/jflow_cli.py rnaseqproc --sample reads-1=myfile_r1.fastq.gz (reads-2=myfile_r2.fastq.gz) --reference-genome reference.fasta (--indexed-genome) --gtf-file model.gtf --protocol (illumina_stranded, other) default: illumina_stranded Other parameters: --trim-reads: to trim reads default: TruSeq Adapter --use-bowtie2: use bowtie2 instead of default bwa --multi-map: If specified, multi-mapping reads/fragments will be counted
 Case 1: you have the transcriptome: --transcriptome : transcriptome fasta file Case 2: you don t have the transcriptome: --reference-genome reference.")
64 Rna-Seq Workflows on Jflow Launch workflow: /usr/local/bioinfo/src/jflow/jflow/bin/jflow_cli.p y rnaseqnoalign --sample reads-1=myfile_r1.fastq.gz (reads-2=myfile_r2.fastq.gz) Case 1: you have the transcriptome: --transcriptome : transcriptome fasta file Case 2: you don t have the transcriptome: --reference-genome reference.fasta --gtf-file model.gtf Rna-Seq Workflows on Jflow The documentation is here: /usr/local/bioinfo/src/jflow/jflow/workflows/rnaseq/doc and give the hidden parameters. In development: A log file containing: the list of commands launched (to have the parameters) and version of the software. Useful references Experimental design: Liu et al., RNA-seq differential expression studies: more sequence or more replication?, 2014, Bioinformatics, Vol. 30 no , pages Schurch et al., How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?, 2016, RNA 22: Pipeline / cufflinks / RSEM: Djebali et al., Bioinformatics pipeline for transcriptome sequencing analysis, Methods in Molecular Biology, 2017, vol Tools / pipelines benchmarks for differentially expressed genes identification: Williams et al., Empirical assessment of analysis workflows for differential expression analysis of human samples using RNA-Seq, BMC bioinformatics, 2017, 18:38. Baruzzo et al., Simulation-based comprehensive benchmarking of RNA-seq aligners, 2017, Nature methods, vol. 14 n 2.
RNA-Seq data analysis
 RNA-Seq data analysis Material Slides: http://genoweb.toulouse.inra.fr/~formation/lignecmd/rnaseq/doc/ pdf : one per page pdf : three per page with comment lines Memento: http://genoweb.toulouse.inra.fr/~formation/lignecmd/rnaseq/doc/mementounix.pdf
RNA-Seq data analysis Material Slides: http://genoweb.toulouse.inra.fr/~formation/lignecmd/rnaseq/doc/ pdf : one per page pdf : three per page with comment lines Memento: http://genoweb.toulouse.inra.fr/~formation/lignecmd/rnaseq/doc/mementounix.pdf
10/06/2014. RNA-Seq analysis. With reference assembly. Cormier Alexandre, PhD student UMR8227, Algal Genetics Group
 RNA-Seq analysis With reference assembly Cormier Alexandre, PhD student UMR8227, Algal Genetics Group Summary 2 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference
RNA-Seq analysis With reference assembly Cormier Alexandre, PhD student UMR8227, Algal Genetics Group Summary 2 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference
RNA-Seq de novo assembly training
 RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Gene Expression analysis with RNA-Seq data
 Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq with the Tuxedo Suite
 RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
Introduction to RNA-Seq. David Wood Winter School in Mathematics and Computational Biology July 1, 2013
 Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12
 Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
RNA-Seq Analysis. Simon Andrews, Laura v
 RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
How to deal with your RNA-seq data?
 How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
How to deal with your RNA-seq data? Rachel Legendre, Thibault Dayris, Adrien Pain, Claire Toffano-Nioche, Hugo Varet École de bioinformatique AVIESAN-IFB 2017 1 Rachel Legendre Bioinformatics 27/11/2018
RNA-Seq Module 2 From QC to differential gene expression.
 RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
RNAseq Differential Gene Expression Analysis Report
 RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
RNA Seq: Methods and Applica6ons. Prat Thiru
 RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
Sequence Analysis 2RNA-Seq
 Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Transcriptomics analysis with RNA seq: an overview Frederik Coppens
 Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Short Read Alignment to a Reference Genome
 Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ),
,") Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
ChIP-seq and RNA-seq. Farhat Habib
 ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq
 ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
Analysis of RNA-seq Data. Feb 8, 2017 Peikai CHEN (PHD)
") Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Transcriptome Assembly, Functional Annotation (and a few other related thoughts)
") Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
RNA-Seq Workshop AChemS Sunil K Sukumaran Monell Chemical Senses Center Philadelphia
 RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Quantifying gene expression
 Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Analysis Datasheet Exosome RNA-seq Analysis
 Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
Deep Sequencing technologies
 Deep Sequencing technologies Gabriela Salinas 30 October 2017 Transcriptome and Genome Analysis Laboratory http://www.uni-bc.gwdg.de/index.php?id=709 Microarray and Deep-Sequencing Core Facility University
Deep Sequencing technologies Gabriela Salinas 30 October 2017 Transcriptome and Genome Analysis Laboratory http://www.uni-bc.gwdg.de/index.php?id=709 Microarray and Deep-Sequencing Core Facility University
Wheat CAP Gene Expression with RNA-Seq
 Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
Wheat CAP Gene Expression with RNA-Seq July 9 th -13 th, 2018 Overview of the workshop, Alina Akhunova http://www.ksre.k-state.edu/igenomics/workshops/ RNA-Seq Workshop Activities Lectures Laboratory Molecular
VM origin. Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE
 X2go: LXDE") VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data
 Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data Jan Hellemans 7th international qpcr & NGS Event - Freising March 24 th, 2015 Therapeutics lncrna oncology
Benchmarking of RNA-seq data processing pipelines using whole transcriptome qpcr expression data Jan Hellemans 7th international qpcr & NGS Event - Freising March 24 th, 2015 Therapeutics lncrna oncology
measuring gene expression December 5, 2017
 measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNAseq Applications in Genome Studies. Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford
 RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Introduction of RNA-Seq Analysis
 Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
RNA standards v May
 Standards, Guidelines and Best Practices for RNA-Seq: 2010/2011 I. Introduction: Sequence based assays of transcriptomes (RNA-seq) are in wide use because of their favorable properties for quantification,
Standards, Guidelines and Best Practices for RNA-Seq: 2010/2011 I. Introduction: Sequence based assays of transcriptomes (RNA-seq) are in wide use because of their favorable properties for quantification,
L3: Short Read Alignment to a Reference Genome
 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
RNA-SEQUENCING ANALYSIS
 RNA-SEQUENCING ANALYSIS Joseph Powell SISG- 2018 CONTENTS Introduction to RNA sequencing Data structure Analyses Transcript counting Alternative splicing Allele specific expression Discovery APPLICATIONS
RNA-SEQUENCING ANALYSIS Joseph Powell SISG- 2018 CONTENTS Introduction to RNA sequencing Data structure Analyses Transcript counting Alternative splicing Allele specific expression Discovery APPLICATIONS
Mapping Next Generation Sequence Reads. Bingbing Yuan Dec. 2, 2010
 Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Sequencing applications. Today's outline. Hands-on exercises. Applications of short-read sequencing: RNA-Seq and ChIP-Seq
 Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Alignment. J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
 Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Applications of short-read
 Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
C3BI. VARIANTS CALLING November Pierre Lechat Stéphane Descorps-Declère
 C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
02 Agenda Item 03 Agenda Item
 01 Agenda Item 02 Agenda Item 03 Agenda Item SOLiD 3 System: Applications Overview April 12th, 2010 Jennifer Stover Field Application Specialist - SOLiD Applications Workflow for SOLiD Application Application
01 Agenda Item 02 Agenda Item 03 Agenda Item SOLiD 3 System: Applications Overview April 12th, 2010 Jennifer Stover Field Application Specialist - SOLiD Applications Workflow for SOLiD Application Application
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Introduction to transcriptome analysis using High Throughput Sequencing technologies. D. Puthier 2012
 Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Systematic evaluation of spliced alignment programs for RNA- seq data
 Systematic evaluation of spliced alignment programs for RNA- seq data Pär G. Engström, Tamara Steijger, Botond Sipos, Gregory R. Grant, André Kahles, RGASP Consortium, Gunnar Rätsch, Nick Goldman, Tim
Systematic evaluation of spliced alignment programs for RNA- seq data Pär G. Engström, Tamara Steijger, Botond Sipos, Gregory R. Grant, André Kahles, RGASP Consortium, Gunnar Rätsch, Nick Goldman, Tim
Alignment & Variant Discovery. J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014
 Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Bioinformatics Monthly Workshop Series. Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory
 Bioinformatics Monthly Workshop Series Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory Schedule for Fall, 2015 PILM Bioinformatics Web Server (09/21/2015)
Bioinformatics Monthly Workshop Series Speaker: Fan Gao, Ph.D Bioinformatics Resource Office The Picower Institute for Learning and Memory Schedule for Fall, 2015 PILM Bioinformatics Web Server (09/21/2015)
RNA-sequencing. Next Generation sequencing analysis Anne-Mette Bjerregaard. Center for biological sequence analysis (CBS)
") RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
Statistical Genomics and Bioinformatics Workshop. Genetic Association and RNA-Seq Studies
 Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
NGS Data Analysis and Galaxy
 NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
Introduction to RNA-Seq in GeneSpring NGS Software
 Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
BST 226 Statistical Methods for Bioinformatics David M. Rocke. March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 3 QC of aligned reads
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 3 QC of aligned reads
Zika infected human samples
 Lecture 16 RNA-seq Zika infected human samples Experimental design ZIKV-infected hnpcs 56 hours after ZIKA and mock infection in parallel cultures were used for global transcriptome analysis. RNA-seq libraries
Lecture 16 RNA-seq Zika infected human samples Experimental design ZIKV-infected hnpcs 56 hours after ZIKA and mock infection in parallel cultures were used for global transcriptome analysis. RNA-seq libraries
measuring gene expression December 11, 2018
 measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and
measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, March 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 1: Introduction into high-throughput
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, March 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 1: Introduction into high-throughput
Consensus Ensemble Approaches Improve De Novo Transcriptome Assemblies
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
RNA-Seq data analysis course September 7-9, 2015
 RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
Analysis of RNA-seq Data
 Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
NGS in Pathology Webinar
 NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
Introduction to NGS analyses
 Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome
 SOFTWARE RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome Bo Li 1 and Colin N Dewey 1,2* Open Access Abstract Background: RNA-Seq is revolutionizing the way
SOFTWARE RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome Bo Li 1 and Colin N Dewey 1,2* Open Access Abstract Background: RNA-Seq is revolutionizing the way
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
RNA-Seq analysis workshop
 RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
Next Generation Sequencing
 Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
Next Generation Sequencing Complete Report Catalogue # and Service: IR16001 rrna depletion (human, mouse, or rat) IR11081 Total RNA Sequencing (80 million reads, 2x75 bp PE) Xxxxxxx - xxxxxxxxxxxxxxxxxxxxxx
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
RNA Sequencing: Experimental Planning and Data Analysis. Nadia Atallah September 12, 2018
 RNA Sequencing: Experimental Planning and Data Analysis Nadia Atallah September 12, 2018 A Next Generation Sequencing (NGS) Refresher Became commercially available in 2005 Construction of a sequencing
RNA Sequencing: Experimental Planning and Data Analysis Nadia Atallah September 12, 2018 A Next Generation Sequencing (NGS) Refresher Became commercially available in 2005 Construction of a sequencing
Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA. March 2, Steven R. Kain, Ph.D. ABRF 2013
 Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA March 2, 2013 Steven R. Kain, Ph.D. ABRF 2013 NuGEN s Core Technologies Selective Sequence Priming Nucleic Acid Amplification
Integrated NGS Sample Preparation Solutions for Limiting Amounts of RNA and DNA March 2, 2013 Steven R. Kain, Ph.D. ABRF 2013 NuGEN s Core Technologies Selective Sequence Priming Nucleic Acid Amplification
Introduction to RNAseq Analysis. Milena Kraus Apr 18, 2016
 Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
An introduction to RNA-seq. Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy
 An introduction to RNA-seq Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy The central dogma Genome = all DNA in an organism (genotype) Transcriptome = all RNA (molecular
An introduction to RNA-seq Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy The central dogma Genome = all DNA in an organism (genotype) Transcriptome = all RNA (molecular
Incorporating Molecular ID Technology. Accel-NGS 2S MID Indexing Kits
 Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
RNA Sequencing. Next gen insight into transcriptomes , Elio Schijlen
 RNA Sequencing Next gen insight into transcriptomes 05-06-2013, Elio Schijlen Transcriptome complete set of transcripts in a cell, and their quantity, for a specific developmental stage or physiological
RNA Sequencing Next gen insight into transcriptomes 05-06-2013, Elio Schijlen Transcriptome complete set of transcripts in a cell, and their quantity, for a specific developmental stage or physiological
QIAseq Targeted Panel Analysis Plugin USER MANUAL
 QIAseq Targeted Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted Panel Analysis 1.1 Windows, macos and Linux June 18, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej
QIAseq Targeted Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted Panel Analysis 1.1 Windows, macos and Linux June 18, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej
Post-assembly Data Analysis
 Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA
 TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA Stranded, Illumina ready library construction in
TECH NOTE Pushing the Limit: A Complete Solution for Generating Stranded RNA Seq Libraries from Picogram Inputs of Total Mammalian RNA Stranded, Illumina ready library construction in
Eucalyptus gene assembly
 Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
High Throughput Sequencing the Multi-Tool of Life Sciences. Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center
 High Throughput Sequencing the Multi-Tool of Life Sciences Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina Still-imaging of clusters (~1000
High Throughput Sequencing the Multi-Tool of Life Sciences Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina Still-imaging of clusters (~1000
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS
 SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
Gene Identification in silico
 Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory http://collaboratory.lifesci.ucla.edu Workshop Outline ü Day 1 UCLA galaxy
Accurate, Fast, and Model-Aware Transcript Expression Quantification
 Accurate, Fast, and Model-Aware Transcript Expression Quantification Carl Kingsford Associate Professor, Computational Biology Department Joint work with Rob Patro & Geet Duggal Challenge of Large-Scale
Accurate, Fast, and Model-Aware Transcript Expression Quantification Carl Kingsford Associate Professor, Computational Biology Department Joint work with Rob Patro & Geet Duggal Challenge of Large-Scale
GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment
 GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment Zhaojun Zhang, Shunping Huang, Jack Wang, Xiang Zhang, Fernando Pardo
GeneScissors: a comprehensive approach to detecting and correcting spurious transcriptome inference owing to RNA-seq reads misalignment Zhaojun Zhang, Shunping Huang, Jack Wang, Xiang Zhang, Fernando Pardo
Genome 373: Mapping Short Sequence Reads II. Doug Fowler
 Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Why QC? Next-Generation Sequencing: Quality Control. Illumina data format. Fastq format:
 Why QC? Next-Generation Sequencing: Quality Control BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute Do you want to include the reads with low quality base calls?
Why QC? Next-Generation Sequencing: Quality Control BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute Do you want to include the reads with low quality base calls?
Bionano Access v1.1 Release Notes
 Bionano Access v1.1 Release Notes Document Number: 30188 Document Revision: C For Research Use Only. Not for use in diagnostic procedures. Copyright 2017 Bionano Genomics, Inc. All Rights Reserved. Table
Bionano Access v1.1 Release Notes Document Number: 30188 Document Revision: C For Research Use Only. Not for use in diagnostic procedures. Copyright 2017 Bionano Genomics, Inc. All Rights Reserved. Table
Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction
 Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction Gunnar Rätsch Friedrich Miescher Laboratory Max Planck Society, Tübingen, Germany NGS Bioinformatics Meeting, Paris (March 24, 2010)
Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction Gunnar Rätsch Friedrich Miescher Laboratory Max Planck Society, Tübingen, Germany NGS Bioinformatics Meeting, Paris (March 24, 2010)
RNAseq and Variant discovery
 RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. GEP goals: Evidence Based Annotation. Evidence for Gene Models 12/26/2018
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Genome annotation & EST
 Genome annotation & EST What is genome annotation? The process of taking the raw DNA sequence produced by the genome sequence projects and adding the layers of analysis and interpretation necessary
Genome annotation & EST What is genome annotation? The process of taking the raw DNA sequence produced by the genome sequence projects and adding the layers of analysis and interpretation necessary
UAB DNA-Seq Analysis Workshop. John Osborne Research Associate Centers for Clinical and Translational Science
 + UAB DNA-Seq Analysis Workshop John Osborne Research Associate Centers for Clinical and Translational Science ozborn@uab.,edu + Thanks in advance You are the Guinea pigs for this workshop! At this point
+ UAB DNA-Seq Analysis Workshop John Osborne Research Associate Centers for Clinical and Translational Science ozborn@uab.,edu + Thanks in advance You are the Guinea pigs for this workshop! At this point
Next-Generation Sequencing: Quality Control
 Next-Generation Sequencing: Quality Control Bingbing Yuan BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Why QC? Do you want to
Next-Generation Sequencing: Quality Control Bingbing Yuan BaRC Hot Topics January 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Why QC? Do you want to
Single-Cell Whole Transcriptome Profiling With the SOLiD. System
 APPLICATION NOTE Single-Cell Whole Transcriptome Profiling Single-Cell Whole Transcriptome Profiling With the SOLiD System Introduction The ability to study the expression patterns of an individual cell
APPLICATION NOTE Single-Cell Whole Transcriptome Profiling Single-Cell Whole Transcriptome Profiling With the SOLiD System Introduction The ability to study the expression patterns of an individual cell