Traditional approaches to 3-D structure determination
|
|
|
- Hilary Armstrong
- 6 years ago
- Views:
Transcription

1 Homology Modeling: A Brief Introduction W. Ross Ellington Biology-IMB Traditional approaches to 3-D structure determination X-ray crystallography: Sufficient protein? Natural protein is often heterogeneous Can it be expressed? Requires full length expression construct If so, in soluble state? Often recombinant protein is present as insoluble inclusion bodies Will it crystallize? Perhaps the most time-consuming task Will the crystals be of diffraction quality? Some crystals will simply not diffract How do you solve the phase problem? heavy metals, selenomethionine, molecular replacement 1
2 NMR spectroscopy: Same problems above with respect to protein availability. Protein must be N 15 and/or C 13 and/or H 2 labeled (bacteria must grow in isotopically enriched media; samples may cost > $2,000 to prepare). Protein must be stable for weeks. Restriction is terms of size of protein (< kd). All methods are time and labor intensive but yield atomic level resolution ( Å) 2
3 Proliferation of deduced amino acid sequences 1. RTPCR PCR amplification of RT products using redundant gene specific primers (very rapid; generates large amounts of data) 2. cdna libraries Cloning of RT products; when screened yield large numbers of cdna sequences. 3. Genomic DNA sequencing efforts Eukaryotes- (man, mouse, chicken, pufferfish, zebrafish, Ciona [tunicate], sea urchin, fruit fly, C. elegans (nematode) and many unicellular pathogenic species) Gene finder software extracts EXONs and assembles ORFs. Prokaryotes- > 25 genomes have been sequenced 3
4 Human genome codes for >30,000 proteins! Convert sequences to 3-D structure > 40% sequence identity- HOMOLOGY MODELING (actually extends to much lower degrees of identity) 20-40% sequence identity- THREADING <25% sequence identity- AB INITIO modeling 4
5 Homology modeling- assume query sequence and homologs share the same basic folds; approach is to pick out the best template from a suite of homologs Homologous proteins- share a common evolutionary origin [ancestor] Orthologs- the same gene present in different species (ca., hemoglobin in fish vs. camels) Paralogs- products of gene duplication events (brain, muscle, sarcomeric mitochondrial and ubiquitous mitochondrial creatine kinases in man) The potential for homology modeling > 500,000 protein sequences are known There are >10,000 experimentally determined 3-D structures of proteins 33% of known protein sequences have similarities to a proteins for which the 3-D structure has been determined Therefore, there is the potential for homology modeling of > 150,000 proteins 5
6 Find homologs 1. Pairwise comparison: compare target sequence individually with other sequences in a database of sequences; FASTA; BLAST Easy to use, on-line sites; probe with protein (BLASTp, tblastn) or nucleotides (BLASTn) NCBI - EMBhttp:// html Expasy - 6
7 2. Psi-Blast: constructs multiple sequence alignments of many sequences which are iteratively sampled from a database; position specific scoring matrix is used to sample database for additional homologs; very useful when percent identity is low NCBS, National Center for Biological Sciences, Bangalore D template matching: pairwise comparison of the query sequence and a protein of known structure; structuredependent scoring function is used to ascertain whether a query protein adopts or not any one of the library of 3D folds; useful when pairwise or multiple sequence approaches have not identified homologs Imperial College 3D PSSM 7
8 4. PROPSEARCH uses the amino acid composition instead. In addition, other properties like molecular weight, content of bulky residues, content of small residues, average hydrophobicity, average charge a.s.o. and the content of selected dipeptide-groups are calculated from the sequence as well. 144 such properties are weighted individually and are used as query vector. The weights are then trained on a set of protein families with known structures, using a genetic algorithm. Sequences in the database are transformed into vectors as well, and the euclidian distance between the query and database sequences is calculated. Distances are rank ordered, and sequences with lowest distance are reported on top. (University of Montpelier) n.html Template choice 1. Higher the sequence identity, the more likely the template will be suitable 2. Most closely related from a phylogenetic point of view 3. Template environment (solvent, ph, temperature, quaternary structure) 4. Quality of the template structure (resolution and R factor) 8
9 Quality: 645 Length: 374 Ratio: Gaps: 5 Percent Similarity: Percent Identity: Match display thresholds for the alignment(s): = IDENTITY : = 2. = 1 Alignment of target (query) with template- using CLUSTAL, GAP (GCG), BLAST (NCBI) etc with manual adjustments UrechiscaupoLK.txt x limulusak.pep October 29, : MAFQNDAKANF...PDYANHGCVVGRHLNFEMYQRLFGKKTAHGVTV 44.. :.... : :.: :. 1 MVDQATLDKLEAGFKKLQEASDCKSLLKKHLTKDVFDSIKNKKTGMGATL DKVIQPSVDNFGNCIGLIAGDEESYEVFKELFDAVINEKHKGFGPNDSQP 94 :. : : :.: 51 LDVIQSGVENLDSGVGIYAPDAESYRTFGPLFDPIIDDYHGGFKLTDKHP APDL.DASKLVGGQFDEKYVKSCRIRTGRGIRGLCYPPSCTRGERREVER 143 :..:: : :. : : :. 101 PKEWGDINTLVDLDPGGQFIISTRVRCGRSLQGYPFNPCLTAEQYKEMEE VITTALAGLSGDLSGTYYPLSKMTPEQENQLIADHFLFQKPTGHLMVNSA 193 :... : :.. : KVSSTLSSMEDELKGTYYPLTGMSKATQQQLIDDHFLFKEGDRFLQTANA SVRDWPDARGIWHNNEKTFLIWINEEDHMRVISMQKGGNVKAVFERFGRG : : : :.. :. 201.CRYWPTGRGIFHNDAKTFLVWVNEEDHLRIISMQKGGDLKTVYKRLVTA LNAIAEQMKKNGREYMWNQRLGYLCACPSNLGTGLRASVHVQLHQLSKHP : :. :. : : VDNIESKL...PFSHDDRFGFLTFCPTNLGTTMRASVHIQLPKLAKDR K.FEDIVVALQLQKRGTGGEHTAAVDDVYDISNAARLKKSEREFVQLLID :. : 295 KVLEDIASKFNLQVRGTRGEHTESEGGVYDISNKRRLGLTEYQAVREMQD GVKKLIDMEQALEAGKSIDDLIPA 366 :.:. 345 GILEMIKMEKAAA Look for highly conserved regionsthis helps with the manual adjustments of alignments! MSTSQNK-IFNQGEDCKPSWTSAGADYNEKDTHLEIMGKPVMERWETPYMHLLATIASSK MTAVAGQPIFGSGEDCKPAWTDASADYDATDEHLEIMGKPVMERWETPYMHKLAQVASCK --MSAAQPIFSKGENCKQVWHDANADYNAADTHLEIMGKPVMERWETPYMHSLATVAASK -MTSSAQPIFSKGEDCKPTWHDANAGYNEADTHXEIMGKPVMERWETPYMHSLATVAASK --MTTAQPIFSKGEDCKAGWHDAGAGYNETDTHLEILGKPVMERWETPYMHSLAIVASSK --MSSDK-IFAEGESCKSAWHDATAGYDETDTHLEIMGKPVMERWETPYMHSLATVAASK -SSSAASPLFAPGEDCGPAWRAAPAAYDTSDTHLQILGKPVMERWETPYMHSLAAAAASR. :* **.* * * * *: * * :*:************** ** *:.: 9
10 Quality: 1415 Length: 419 Ratio: Gaps: 2 Percent Similarity: Percent Identity: Match display thresholds for the alignment(s): = IDENTITY : = 2. = 1 EXAMPLE- Chaetopterus variopedatus (polychaete= marine worm) mitochondrial creatine kinase vs. chicken sarcomeric MiCK (X-ray crystal structure published in 1995 by Fritz-Wolf et al.; PDB- 1crk) correctchaetmick.txt x chickenmicksar.pep February 25, : MRLGTSNVHLRYPA 14.: : 1 MAGTFGRLLAGRVTAALFAAAGSGVLTTGYLLNQQNVKATVHEKRKLFPP SANYPDLSQHNNIMASNLTPVIYAKLRDKVTPNGVTLNLCIQTGVDNPGH SADYPDLRKHNNCMAECLTPAIYAKLRDKLTPNGYSLDQCIQTGVDNPGH PFIKTVGLVAGDEESYEVFADLFDKCIDERHGGYKPWD.KHPTDLDSTKL 113 : ::.. : 101 PFIKTVGMVAGDEESYEVFAEIFDPVIKARHNGYDPRTMKHHTDLDASKI RGGNFDPKYVLSSRVRTGRCIRGLSLPPACSRAERREVERVVVEALNGLQ 163 :. 151 THGQFDERYVLSSRVRTGRSIRGLSLPPACSRAERREVENVVVTALAGLK GDLAGKYFPLAKMTDAEQEKLIEDHFLFDKPVSPLLLASGMARDWPDARG 213. :.: : :. :. 201 GDLSGKYYSLTNMSERDQQQLIDDHFLFDKPVSPLLTCAGMARDWPDARG IWHNDKKNFLVWVNEEDHTRVISMQKGGNMREVFDRFCNGLQKVENLIQS 263. : : : : IWHNNDKTFLVWINEEDHTRVISMEKGGNMKRVFERFCRGLKEVERLIKE RGWEFMWNEHLGYVLTCPSNLGTGLRAGVHIKIPKLAKDPRLNEVLKKMN 313 : : :..:. : 301 RGWEFMWNERLGYVLTCPSNLGTGLRAGVHVKLPRLSKDPRFPKILENLR LQKRGTGGVDTAATGDTFDISNSDRLGKSEVQLVQQVVDGIDLLIQMEKR 363 : : : : : :. : : 351 LQKRGTGGVDTAAVADVYDISNLDRMGRSEVELVQIVIDGVNYLVDCEKK LERGQ..RIDDLIPK : :: :. 401 LEKGQDIKVPPPLPQFGRK 419 Generate coordinates for SCRs and VRs using template(s0 Web-based 1. Swiss Model- Glaxo Welcome 2. What If- EMBL Programs 1. Modeller- free; Unix, Linux [Rockefeller U.] 2. Insight II ( homology )- Unix [MSI] 10
11 SwissModel- What If- Modeller- Insight IIhttp:// PDB file-header HEADER TRANSFERASE 08-MAR-96 1CRK Background information TITLE MITOCHONDRIAL CREATINE KINASE COMPND MOL_ID: 1; COMPND 2 MOLECULE: CREATINE KINASE; COMPND 3 CHAIN: A, B, C, D; COMPND 4 EC: ; COMPND 5 BIOLOGICAL_UNIT: OCTAMER SOURCE MOL_ID: 1; SOURCE SOURCE SOURCE SOURCE SOURCE SOURCE KEYWDS EXPDTA AUTHOR 2 ORGANISM_SCIENTIFIC: GALLUS GALLUS; 3 ORGANISM_COMMON: CHICKEN; 4 ORGAN: HEART; 5 TISSUE: MUSCLE; 6 CELL: SARCOMER; 7 ORGANELLE: MITOCHONDRIA TRANSFERASE, CREATINE KINASE X-RAY DIFFRACTION K.FRITZ-WOLF,T.SCHNYDER,T.WALLIMANN,W.KABSCH REVDAT 1 07-JUL-97 1CRK 0 JRNL AUTH K.FRITZ-WOLF,T.SCHNYDER,T.WALLIMANN,W.KABSCH JRNL TITL STRUCTURE OF MITOCHONDRIAL CREATINE KINASE JRNL REF NATURE V JRNL REFN ASTM NATUAS UK ISSN
12 PDB file-remarks REMARK 1 Properties of the structure like resolution, error etc. REMARK 2 REMARK 2 RESOLUTION. 3.0 ANGSTROMS. REMARK 3 REMARK 3 REFINEMENT. REMARK 3 PROGRAM : X-PLOR REMARK 3 AUTHORS : BRUNGER REMARK 3 REMARK 3 DATA USED IN REFINEMENT. REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 3. REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : 8. REMARK 3 DATA CUTOFF (SIGMA(F)) : 0. REMARK 3 DATA CUTOFF HIGH (ABS(F)) : NULL REMARK 3 DATA CUTOFF LOW (ABS(F)) : NULL REMARK 3 COMPLETENESS (WORKING+TEST) (%) : 99.6 REMARK 3 NUMBER OF REFLECTIONS : REMARK 3 REMARK 3 FIT TO DATA USED IN REFINEMENT. REMARK 3 CROSS-VALIDATION METHOD : NULL REMARK 3 FREE R VALUE TEST SET SELECTION : NULL REMARK 3 R VALUE (WORKING SET) : REMARK 3 FREE R VALUE : REMARK 3 FREE R VALUE TEST SET SIZE (%) : 10. REMARK 3 FREE R VALUE TEST SET COUNT : NULL REMARK 3 ESTIMATED ERROR OF FREE R VALUE : NULL PDB file- helix/sheet HELIX ALA D 108 LYS D HELIX ARG D 143 GLY D HELIX GLY D 162 LEU D HELIX GLU D 176 ASP D HELIX PRO D 195 ALA D HELIX MET D 241 ARG D HELIX PRO D 279 ASN D HELIX PRO D 295 LYS D HELIX PHE D 303 LEU D HELIX GLU D 341 GLU D SHEET 1 A 8 GLY A 166 SER A SHEET 2 A 8 GLY A 211 ASN A N HIS A 214 O LYS A 167 SHEET 3 A 8 PHE A 220 ILE A N ILE A 224 O GLY A 211 SHEET 4 A 8 THR A 230 LYS A N ILE A 233 O LEU A 221 SHEET 5 A 8 VAL A 121 ARG A N ARG A 130 O THR A 230 Secondary structure- a helices and b sheets 12
13 PDB file- atomic coordinates of atoms ATOM 2 CA THR A C ATOM 3 C THR A C ATOM 4 O THR A O ATOM 5 CB THR A C ATOM 6 OG1 THR A O ATOM 7 CG2 THR A C ATOM 8 N VAL A N ATOM 9 CA VAL A C ATOM 10 C VAL A C ATOM 11 O VAL A O ATOM 12 CB VAL A C ATOM 13 CG1 VAL A C ATOM 14 CG2 VAL A C ATOM 15 N HIS A N ATOM 16 CA HIS A C ATOM 17 C HIS A C ATOM 18 O HIS A O ATOM 19 CB HIS A C ATOM 20 CG HIS A C ATOM 21 ND1 HIS A N ATOM 22 CD2 HIS A C ATOM 23 CE1 HIS A C ATOM 24 NE2 HIS A N Three general methods for initial model construction 1. Assembly of rigid bodies- backbone of target is arranged to template positions of a carbons RAPPER (Cambridge University) is a discrete conformational sampling algorithm for restraint-based protein modelling. It has been used for all-atom loop modelling, whole protein modelling under limited (Calpha) restraints, comparative modelling, ab initio structure prediction, structure validation, and experimental structure determination with X-ray and nuclear magnetic resonance spectroscopy 13
14 2. Segment matching (coordinate reconstruction)- most hexapeptide segments of protein structure can be clustered into approximately 100 structural classes; subset of atomic positions in template are used as guiding positions to fit segments from the target into these positions; can be used to model main chain and side chain atoms SegMod component available in UCLA s Genemine 3. Satisfaction of spatial restraints-model is built on minimizing violations of spatial restraints (constraints) as defined from the template; homology derived restraints (mainchain and sidechain dihedrals, mainchain CA-CA distances, sidechainmainchain distances etc) and stereochemical (bond angles, dihedral angles, non-bonded atom-atom contacts) (MODELLER) A. Sali & T.L. Blundell. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, , A. Fiser, R.K. Do, & A. Sali. Modeling of loops in protein structures, Protein Science ,
15 Initial model will yield a framework of SCRs consisting of the following: 1. Backbone of target protein is arranged to a carbons of the template. 2. Secondary structural elements are present 3. F and Y angles are arranged in SCRs Initial modeling will yield a framework which lacks two important components- (a) VRs [nonconserved loops] and (b) side chains. Quality: 1415 Length: 419 Ratio: Gaps: 2 Percent Similarity: Percent Identity: Match display thresholds for the alignment(s): = IDENTITY : = 2. = 1 correctchaetmick.txt x chickenmicksar.pep February 25, : MRLGTSNVHLRYPA 14.: : 1 MAGTFGRLLAGRVTAALFAAAGSGVLTTGYLLNQQNVKATVHEKRKLFPP SANYPDLSQHNNIMASNLTPVIYAKLRDKVTPNGVTLNLCIQTGVDNPGH SADYPDLRKHNNCMAECLTPAIYAKLRDKLTPNGYSLDQCIQTGVDNPGH PFIKTVGLVAGDEESYEVFADLFDKCIDERHGGYKPWD.KHPTDLDSTKL 113 : ::.. : 101 PFIKTVGMVAGDEESYEVFAEIFDPVIKARHNGYDPRTMKHHTDLDASKI RGGNFDPKYVLSSRVRTGRCIRGLSLPPACSRAERREVERVVVEALNGLQ 163 :. 151 THGQFDERYVLSSRVRTGRSIRGLSLPPACSRAERREVENVVVTALAGLK GDLAGKYFPLAKMTDAEQEKLIEDHFLFDKPVSPLLLASGMARDWPDARG 213. :.: : :. :. 201 GDLSGKYYSLTNMSERDQQQLIDDHFLFDKPVSPLLTCAGMARDWPDARG IWHNDKKNFLVWVNEEDHTRVISMQKGGNMREVFDRFCNGLQKVENLIQS 263. : : : : IWHNNDKTFLVWINEEDHTRVISMEKGGNMKRVFERFCRGLKEVERLIKE RGWEFMWNEHLGYVLTCPSNLGTGLRAGVHIKIPKLAKDPRLNEVLKKMN 313 : : :..:. : 301 RGWEFMWNERLGYVLTCPSNLGTGLRAGVHVKLPRLSKDPRFPKILENLR LQKRGTGGVDTAATGDTFDISNSDRLGKSEVQLVQQVVDGIDLLIQMEKR 363 : : : : : :. : : 351 LQKRGTGGVDTAAVADVYDISNLDRMGRSEVELVQIVIDGVNYLVDCEKK LERGQ..RIDDLIPK : :: :. 401 LEKGQDIKVPPPLPQFGRK
16 Loop modeling- changes in loops usually occur in exposed regions that connect secondary structure elements. No reliable methods are available for constructing loops longer than 5 residues. Two general approaches: 1. Ab initio loop prediction- conformational search or enumeration of conformations in a given environment guided by a scoring or energy function. Also available on the Cambridge University RAVEN site 2. Database approaches-consists of finding a portion of the main chain which fits the stem regions of a particular loop. The database of protein folds is searched and fitted within the constraints of the loop sequence, stem structure and energy optimization criteria. Loop database at PKUBIOS (China) loop.htm 16
17 Side chains-defined by C 1, C 2. (dihedral angles) -protein side chains play a key role in molecular recognition and packing of hydrophobic cores of globular proteins -significant correlations exist between C 1, C 2 and F and Y angles -side chain conformations exist in a limited number of canonical shapes (rotamers) -rotamer libraries can be constructed where only 3-50 conformations are taken into account for each side chain -approaches involve iterative sampling of rotamers for each side chain into the target backbone with scoring matrices and attention paid to steric clashes of sidechain with mainchain. Refinement by molecular mechanics -restrain the region of the model that is most likely correct and focus on suspect areas or perform on entire molecule -idealize bond geometry and remove unfavorable non-bonded contacts by protein force fields using AMBER Assisted Model Building with Energy Refinement ( CHARMm Chemistry at HARvard Molecular mechanics ( GROMOS Molecular Dynamics ( 17
18 -this essentially is an energy minimization approach; during force field calculations there is a tendency for the structure to drift away from the control structure. This drift can be reduced by -(a) limiting the number of minimization steps and - (b) restraining many of the alpha carbons. Errors in homology models (a) errors in side chain packing- as proteins diverge, the packing of side chains in the core changes, (b) distortions or shifts in correctly aligned regionssequence divergence may result in main chain conformation changes even though overall fold is the same, (c) errors in regions without a template- occurs in segments of a target protein for which there are no equivalents in the template 18
19 (d) errors due to misalignments- most common error and (e) incorrect templates- is a problem that occurs when using distantly related templates. Evaluating models Explicit approach: overlay the model on the template and evaluate rmsd (root mean square deviation of the a carbons) -experimental rmsd values by X-ray crystallography range from from 0.5 Å for the same protein to 1 Å for proteins with > 50% identity -successful model has < 2 Å rmsd from template (if template has a sequence identity > 60% then the above criterion would be met with a success rate > 70%) -sequence identity of target and template is critical 19
20 Judging quality of homology models 1. ACCURACY- how well it fits the templates upon which it was built (rmsd deviation) 2. CONFIDENCE FACTOR (Model B-factor)- SWISS MODEL provides a B-factor which is an uncertainty factor; higher the value the lower the amount of actual structural support is present for a particular portion of the model 3. INCORRECTNESS- presence of hydrophobic groups on the surface or polar groups in the interior that do not have hydrogen bonding or ionic bonding capabilities satisfied by their neighbors or steric clashes or unreasonable structural parameters such as bond angles/lengths. 20


21 4. Ramachandran diagram- plot shows main-chain conformational angles; there are a finite number of F and Y angle combinations as defined in the plot; glycines, which lack side chains, often fall out of allowed regions. Residues other than glycines in a model that are not present in allowed regions are suspect. 5. REASONABLENESS- defined as whether the model is in keeping with expectations for similar proteins. Assessed by summing up the probabilities that each residue should occur in the environment in which it is found in the model. For all PDB models, each of the 20 amino acids has a certain probability of belonging to one of the following classes: solvent-accessible surface, buried polar, exposed nonpolar, helix, sheet or turn. Regions in the model that do not fit expectations are suspect. Threading Energy- a criterion for reasonableness; higher the energy, lower the reasonableness 21
22 Detection of errors 1.manual inspection 2. checking stereochemistry PROCHECK ( WHATCHECK ( SQUID ( 3. statistical analyses of structures based on compiled 3-D features of many proteins VERIFY3D ( ProsaII ( 22

23 23
24 24

25 Quality: 1415 Length: 419 Ratio: Gaps: 2 Percent Similarity: Percent Identity: Match display thresholds for the alignment(s): = IDENTITY : = 2. = 1 correctchaetmick.txt x chickenmicksar.pep February 25, : MRLGTSNVHLRYPA 14.: : 1 MAGTFGRLLAGRVTAALFAAAGSGVLTTGYLLNQQNVKATVHEKRKLFPP SANYPDLSQHNNIMASNLTPVIYAKLRDKVTPNGVTLNLCIQTGVDNPGH SADYPDLRKHNNCMAECLTPAIYAKLRDKLTPNGYSLDQCIQTGVDNPGH PFIKTVGLVAGDEESYEVFADLFDKCIDERHGGYKPWD.KHPTDLDSTKL 113 : ::.. : 101 PFIKTVGMVAGDEESYEVFAEIFDPVIKARHNGYDPRTMKHHTDLDASKI RGGNFDPKYVLSSRVRTGRCIRGLSLPPACSRAERREVERVVVEALNGLQ 163 :. 151 THGQFDERYVLSSRVRTGRSIRGLSLPPACSRAERREVENVVVTALAGLK GDLAGKYFPLAKMTDAEQEKLIEDHFLFDKPVSPLLLASGMARDWPDARG 213. :.: : :. :. 201 GDLSGKYYSLTNMSERDQQQLIDDHFLFDKPVSPLLTCAGMARDWPDARG IWHNDKKNFLVWVNEEDHTRVISMQKGGNMREVFDRFCNGLQKVENLIQS 263. : : : : IWHNNDKTFLVWINEEDHTRVISMEKGGNMKRVFERFCRGLKEVERLIKE RGWEFMWNEHLGYVLTCPSNLGTGLRAGVHIKIPKLAKDPRLNEVLKKMN 313 : : :..:. : 301 RGWEFMWNERLGYVLTCPSNLGTGLRAGVHVKLPRLSKDPRFPKILENLR LQKRGTGGVDTAATGDTFDISNSDRLGKSEVQLVQQVVDGIDLLIQMEKR 363 : : : : : :. : : 351 LQKRGTGGVDTAAVADVYDISNLDRMGRSEVELVQIVIDGVNYLVDCEKK LERGQ..RIDDLIPK : :: :. 401 LEKGQDIKVPPPLPQFGRK 419 Chicken sarmick 25

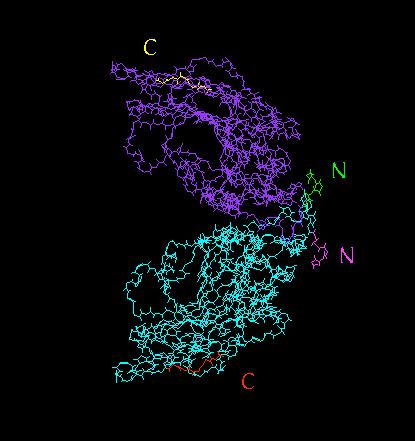
26 26
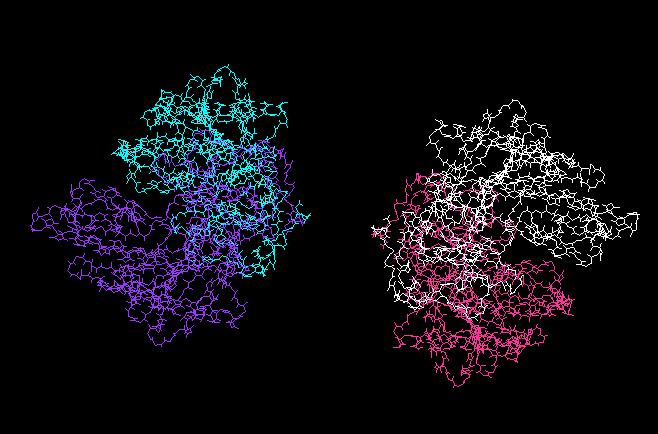

27 27
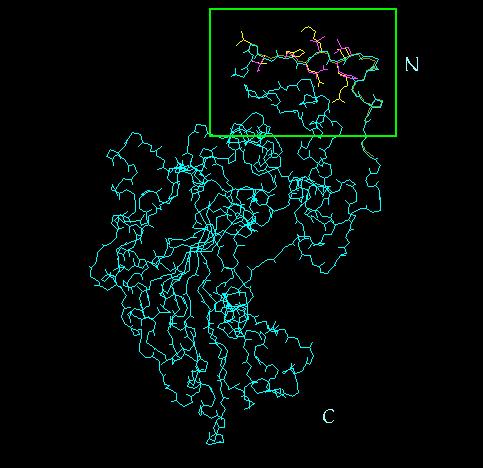

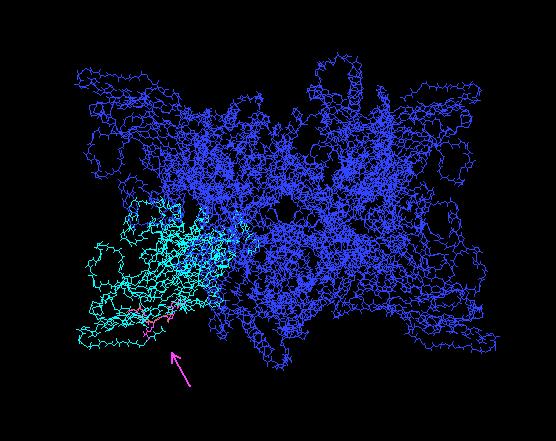
28 28

29 29
30 Where do we go from here? vhigh through-put structure determination efforts will never come close to determining the bulk of structures vhowever, a dictionary of most of the possible folds is a realistic vgiven these folds, homology modeling affords great promise in identification and characterization of structural genes whose products have an unknown function 30
Structural bioinformatics
 Structural bioinformatics Why structures? The representation of the molecules in 3D is more informative New properties of the molecules are revealed, which can not be detected by sequences Eran Eyal Plant
Structural bioinformatics Why structures? The representation of the molecules in 3D is more informative New properties of the molecules are revealed, which can not be detected by sequences Eran Eyal Plant
Virtual bond representation
 Today s subjects: Virtual bond representation Coordination number Contact maps Sidechain packing: is it an instrumental way of selecting and consolidating a fold? ASA of proteins Interatomic distances
Today s subjects: Virtual bond representation Coordination number Contact maps Sidechain packing: is it an instrumental way of selecting and consolidating a fold? ASA of proteins Interatomic distances
Protein 3D Structure Prediction
 Protein 3D Structure Prediction Michael Tress CNIO ?? MREYKLVVLGSGGVGKSALTVQFVQGIFVDE YDPTIEDSYRKQVEVDCQQCMLEILDTAGTE QFTAMRDLYMKNGQGFALVYSITAQSTFNDL QDLREQILRVKDTEDVPMILVGNKCDLEDER VVGKEQGQNLARQWCNCAFLESSAKSKINVN
Protein 3D Structure Prediction Michael Tress CNIO ?? MREYKLVVLGSGGVGKSALTVQFVQGIFVDE YDPTIEDSYRKQVEVDCQQCMLEILDTAGTE QFTAMRDLYMKNGQGFALVYSITAQSTFNDL QDLREQILRVKDTEDVPMILVGNKCDLEDER VVGKEQGQNLARQWCNCAFLESSAKSKINVN
CFSSP: Chou and Fasman Secondary Structure Prediction server
 Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Homology Modelling. Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen
 Homology Modelling Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen Why are Protein Structures so Interesting? They provide a detailed picture of interesting biological features,
Homology Modelling Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen Why are Protein Structures so Interesting? They provide a detailed picture of interesting biological features,
Homology Modelling. Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen
 Homology Modelling Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen Why are Protein Structures so Interesting? They provide a detailed picture of interesting biological features,
Homology Modelling Thomas Holberg Blicher NNF Center for Protein Research University of Copenhagen Why are Protein Structures so Interesting? They provide a detailed picture of interesting biological features,
Why learn sequence database searching? Searching Molecular Databases with BLAST
 Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
Dynamic Programming Algorithms
 Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Structure formation and association of biomolecules. Prof. Dr. Martin Zacharias Lehrstuhl für Molekulardynamik (T38) Technische Universität München
 Technische Universität München") Structure formation and association of biomolecules Prof. Dr. Martin Zacharias Lehrstuhl für Molekulardynamik (T38) Technische Universität München Motivation Many biomolecules are chemically synthesized
Structure formation and association of biomolecules Prof. Dr. Martin Zacharias Lehrstuhl für Molekulardynamik (T38) Technische Universität München Motivation Many biomolecules are chemically synthesized
Zool 3200: Cell Biology Exam 3 3/6/15
 Name: Trask Zool 3200: Cell Biology Exam 3 3/6/15 Answer each of the following questions in the space provided; circle the correct answer or answers for each multiple choice question and circle either
Name: Trask Zool 3200: Cell Biology Exam 3 3/6/15 Answer each of the following questions in the space provided; circle the correct answer or answers for each multiple choice question and circle either
Sequence Databases and database scanning
 Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Outline. Evolution. Adaptive convergence. Common similarity problems. Chapter 7: Similarity searches on sequence databases
 Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Packing of Secondary Structures
 7.88 Lecture Notes - 5 7.24/7.88J/5.48J The Protein Folding and Human Disease Packing of Secondary Structures Packing of Helices against sheets Packing of sheets against sheets Parallel Orthogonal Table:
7.88 Lecture Notes - 5 7.24/7.88J/5.48J The Protein Folding and Human Disease Packing of Secondary Structures Packing of Helices against sheets Packing of sheets against sheets Parallel Orthogonal Table:
MATH 5610, Computational Biology
 MATH 5610, Computational Biology Lecture 2 Intro to Molecular Biology (cont) Stephen Billups University of Colorado at Denver MATH 5610, Computational Biology p.1/24 Announcements Error on syllabus Class
MATH 5610, Computational Biology Lecture 2 Intro to Molecular Biology (cont) Stephen Billups University of Colorado at Denver MATH 5610, Computational Biology p.1/24 Announcements Error on syllabus Class
6-Foot Mini Toober Activity
 Big Idea The interaction between the substrate and enzyme is highly specific. Even a slight change in shape of either the substrate or the enzyme may alter the efficient and selective ability of the enzyme
Big Idea The interaction between the substrate and enzyme is highly specific. Even a slight change in shape of either the substrate or the enzyme may alter the efficient and selective ability of the enzyme
Supporting Online Material. Av1 and Av2 were isolated and purified under anaerobic conditions according to
 Supporting Online Material Materials and Methods Av1 and Av2 were isolated and purified under anaerobic conditions according to published protocols (S1). Crystals of nf-, pcp- and adp-av2:av1 complexes
Supporting Online Material Materials and Methods Av1 and Av2 were isolated and purified under anaerobic conditions according to published protocols (S1). Crystals of nf-, pcp- and adp-av2:av1 complexes
BIOINFORMATICS Introduction
 BIOINFORMATICS Introduction Mark Gerstein, Yale University bioinfo.mbb.yale.edu/mbb452a 1 (c) Mark Gerstein, 1999, Yale, bioinfo.mbb.yale.edu What is Bioinformatics? (Molecular) Bio -informatics One idea
BIOINFORMATICS Introduction Mark Gerstein, Yale University bioinfo.mbb.yale.edu/mbb452a 1 (c) Mark Gerstein, 1999, Yale, bioinfo.mbb.yale.edu What is Bioinformatics? (Molecular) Bio -informatics One idea
Alpha-helices, beta-sheets and U-turns within a protein are stabilized by (hint: two words).
.") 1 Quiz1 Q1 2011 Alpha-helices, beta-sheets and U-turns within a protein are stabilized by (hint: two words) Value Correct Answer 1 noncovalent interactions 100% Equals hydrogen bonds (100%) Equals H-bonds
1 Quiz1 Q1 2011 Alpha-helices, beta-sheets and U-turns within a protein are stabilized by (hint: two words) Value Correct Answer 1 noncovalent interactions 100% Equals hydrogen bonds (100%) Equals H-bonds
Protein Folding Problem I400: Introduction to Bioinformatics
 Protein Folding Problem I400: Introduction to Bioinformatics November 29, 2004 Protein biomolecule, macromolecule more than 50% of the dry weight of cells is proteins polymer of amino acids connected into
Protein Folding Problem I400: Introduction to Bioinformatics November 29, 2004 Protein biomolecule, macromolecule more than 50% of the dry weight of cells is proteins polymer of amino acids connected into
Introduction to protein structure analysis and prediction
 Introduction to protein structure analysis and prediction Mónica Chagoyen monica.chagoyen@cnb.csic.es Protein sequence analysis and prediction service Centro Nacional de Biotecnologia (CNB-CSIC) 24-26
Introduction to protein structure analysis and prediction Mónica Chagoyen monica.chagoyen@cnb.csic.es Protein sequence analysis and prediction service Centro Nacional de Biotecnologia (CNB-CSIC) 24-26
CSE : Computational Issues in Molecular Biology. Lecture 19. Spring 2004
 CSE 397-497: Computational Issues in Molecular Biology Lecture 19 Spring 2004-1- Protein structure Primary structure of protein is determined by number and order of amino acids within polypeptide chain.
CSE 397-497: Computational Issues in Molecular Biology Lecture 19 Spring 2004-1- Protein structure Primary structure of protein is determined by number and order of amino acids within polypeptide chain.
Problem Set Unit The base ratios in the DNA and RNA for an onion (Allium cepa) are given below.
 are given below.") Problem Set Unit 3 Name 1. Which molecule is found in both DNA and RNA? A. Ribose B. Uracil C. Phosphate D. Amino acid 2. Which molecules form the nucleotide marked in the diagram? A. phosphate, deoxyribose
Problem Set Unit 3 Name 1. Which molecule is found in both DNA and RNA? A. Ribose B. Uracil C. Phosphate D. Amino acid 2. Which molecules form the nucleotide marked in the diagram? A. phosphate, deoxyribose
Bioinformatics. ONE Introduction to Biology. Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012
 Bioinformatics ONE Introduction to Biology Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012 Biology Review DNA RNA Proteins Central Dogma Transcription Translation
Bioinformatics ONE Introduction to Biology Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012 Biology Review DNA RNA Proteins Central Dogma Transcription Translation
Chapter 8. One-Dimensional Structural Properties of Proteins in the Coarse-Grained CABS Model. Sebastian Kmiecik and Andrzej Kolinski.
 Chapter 8 One-Dimensional Structural Properties of Proteins in the Coarse-Grained CABS Model Abstract Despite the significant increase in computational power, molecular modeling of protein structure using
Chapter 8 One-Dimensional Structural Properties of Proteins in the Coarse-Grained CABS Model Abstract Despite the significant increase in computational power, molecular modeling of protein structure using
Protein Structure Prediction. christian studer , EPFL
 Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
11 questions for a total of 120 points
 Your Name: BYS 201, Final Exam, May 3, 2010 11 questions for a total of 120 points 1. 25 points Take a close look at these tables of amino acids. Some of them are hydrophilic, some hydrophobic, some positive
Your Name: BYS 201, Final Exam, May 3, 2010 11 questions for a total of 120 points 1. 25 points Take a close look at these tables of amino acids. Some of them are hydrophilic, some hydrophobic, some positive
Introduction to Proteins
 Introduction to Proteins Lecture 4 Module I: Molecular Structure & Metabolism Molecular Cell Biology Core Course (GSND5200) Matthew Neiditch - Room E450U ICPH matthew.neiditch@umdnj.edu What is a protein?
Introduction to Proteins Lecture 4 Module I: Molecular Structure & Metabolism Molecular Cell Biology Core Course (GSND5200) Matthew Neiditch - Room E450U ICPH matthew.neiditch@umdnj.edu What is a protein?
Residue Contact Prediction for Protein Structure using 2-Norm Distances
 Residue Contact Prediction for Protein Structure using 2-Norm Distances Nikita V Mahajan Department of Computer Science &Engg GH Raisoni College of Engineering, Nagpur LGMalik Department of Computer Science
Residue Contact Prediction for Protein Structure using 2-Norm Distances Nikita V Mahajan Department of Computer Science &Engg GH Raisoni College of Engineering, Nagpur LGMalik Department of Computer Science
Protein Sequence Analysis. BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl)
") Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Polypeptide backbone (or the main chain)
") Polypeptide backbone (or the main chain) [IUPA-IUB ommission on Biochemical omenclature, Abbreviations and Symbols for the Description of the onfor mation of Polypeptide hains. Eur. J. Biochem., 969, 7,
Polypeptide backbone (or the main chain) [IUPA-IUB ommission on Biochemical omenclature, Abbreviations and Symbols for the Description of the onfor mation of Polypeptide hains. Eur. J. Biochem., 969, 7,
Why Use BLAST? David Form - August 15,
 Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
Ref: Crystallgraphy made crystal clear, Chapter 6. Preparation of heavy atom derivatives. Heavy-atom data bank:
 Obtaining Phases 1. Phasing by Isomorphous Replacement Methods SIR, Single Isomorphous Replacement method MIR, Multiple Isomorphous Replacement method (MIR) SIRAS, Single Isomorphous Replacement Anomalous
Obtaining Phases 1. Phasing by Isomorphous Replacement Methods SIR, Single Isomorphous Replacement method MIR, Multiple Isomorphous Replacement method (MIR) SIRAS, Single Isomorphous Replacement Anomalous
Fundamentals of Protein Structure
 Outline Fundamentals of Protein Structure Yu (Julie) Chen and Thomas Funkhouser Princeton University CS597A, Fall 2005 Protein structure Primary Secondary Tertiary Quaternary Forces and factors Levels
Outline Fundamentals of Protein Structure Yu (Julie) Chen and Thomas Funkhouser Princeton University CS597A, Fall 2005 Protein structure Primary Secondary Tertiary Quaternary Forces and factors Levels
Computer aided modeling of a fructose repressor
 Computer aided modeling of a fructose repressor Miia Helanto and Kristiina Kiviharju Abstract The bioconversion of fructose to mannitol is a commercially interesting bioprocess. Fructose can, however be
Computer aided modeling of a fructose repressor Miia Helanto and Kristiina Kiviharju Abstract The bioconversion of fructose to mannitol is a commercially interesting bioprocess. Fructose can, however be
Protein NMR II. Lecture 5
 Protein NMR II Lecture 5 Standard and NMR chemical shifts in proteins Residue N A A B O Ala 123.8 4.35 52.5 19.0 177.1 ys 118.8 4.65 58.8 28.6 174.8 Asp 120.4 4.76 54.1 40.8 177.2 Glu 120.2 4.29 56.7 29.7
Protein NMR II Lecture 5 Standard and NMR chemical shifts in proteins Residue N A A B O Ala 123.8 4.35 52.5 19.0 177.1 ys 118.8 4.65 58.8 28.6 174.8 Asp 120.4 4.76 54.1 40.8 177.2 Glu 120.2 4.29 56.7 29.7
Building an Antibody Homology Model
 Application Guide Antibody Modeling: A quick reference guide to building Antibody homology models, and graphical identification and refinement of CDR loops in Discovery Studio. Francisco G. Hernandez-Guzman,
Application Guide Antibody Modeling: A quick reference guide to building Antibody homology models, and graphical identification and refinement of CDR loops in Discovery Studio. Francisco G. Hernandez-Guzman,
3D Structure Prediction with Fold Recognition/Threading. Michael Tress CNB-CSIC, Madrid
 3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
X-ray structures of fructosyl peptide oxidases revealing residues responsible for gating oxygen access in the oxidative half reaction
 X-ray structures of fructosyl peptide oxidases revealing residues responsible for gating oxygen access in the oxidative half reaction Tomohisa Shimasaki 1, Hiromi Yoshida 2, Shigehiro Kamitori 2 & Koji
X-ray structures of fructosyl peptide oxidases revealing residues responsible for gating oxygen access in the oxidative half reaction Tomohisa Shimasaki 1, Hiromi Yoshida 2, Shigehiro Kamitori 2 & Koji
 Problem: The GC base pairs are more stable than AT base pairs. Why? 5. Triple-stranded DNA was first observed in 1957. Scientists later discovered that the formation of triplestranded DNA involves a type
Problem: The GC base pairs are more stable than AT base pairs. Why? 5. Triple-stranded DNA was first observed in 1957. Scientists later discovered that the formation of triplestranded DNA involves a type
Overview. Secondary Structure. Tertiary Structure
 Protein Structure Disclaimer: All information and images were taken from outside sources and the author claims no legal ownership of any material. Sources for images are linked on each slide and the information
Protein Structure Disclaimer: All information and images were taken from outside sources and the author claims no legal ownership of any material. Sources for images are linked on each slide and the information
Protein Bioinformatics Part I: Access to information
 Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
MBMB451A Section1 Fall 2008 KEY These questions may have more than one correct answer
 MBMB451A Section1 Fall 2008 KEY These questions may have more than one correct answer 1. In a double stranded molecule of DNA, the ratio of purines : pyrimidines is (a) variable (b) determined by the base
MBMB451A Section1 Fall 2008 KEY These questions may have more than one correct answer 1. In a double stranded molecule of DNA, the ratio of purines : pyrimidines is (a) variable (b) determined by the base
Basic Bioinformatics: Homology, Sequence Alignment,
 Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Mutation Rates and Sequence Changes
 s and Sequence Changes part of Fortgeschrittene Methoden in der Bioinformatik Computational EvoDevo University Leipzig Leipzig, WS 2011/12 From Molecular to Population Genetics molecular level substitution
s and Sequence Changes part of Fortgeschrittene Methoden in der Bioinformatik Computational EvoDevo University Leipzig Leipzig, WS 2011/12 From Molecular to Population Genetics molecular level substitution
Lecture for Wednesday. Dr. Prince BIOL 1408
 Lecture for Wednesday Dr. Prince BIOL 1408 THE FLOW OF GENETIC INFORMATION FROM DNA TO RNA TO PROTEIN Copyright 2009 Pearson Education, Inc. Genes are expressed as proteins A gene is a segment of DNA that
Lecture for Wednesday Dr. Prince BIOL 1408 THE FLOW OF GENETIC INFORMATION FROM DNA TO RNA TO PROTEIN Copyright 2009 Pearson Education, Inc. Genes are expressed as proteins A gene is a segment of DNA that
Name: TOC#. Data and Observations: Figure 1: Amino Acid Positions in the Hemoglobin of Some Vertebrates
 Name: TOC#. Comparing Primates Background: In The Descent of Man, the English naturalist Charles Darwin formulated the hypothesis that human beings and other primates have a common ancestor. A hypothesis
Name: TOC#. Comparing Primates Background: In The Descent of Man, the English naturalist Charles Darwin formulated the hypothesis that human beings and other primates have a common ancestor. A hypothesis
Sequence Analysis '17 -- lecture Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction
 Sequence Analysis '17 -- lecture 16 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction Alpha helix Right-handed helix. H-bond is from the oxygen at i to the nitrogen
Sequence Analysis '17 -- lecture 16 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction Alpha helix Right-handed helix. H-bond is from the oxygen at i to the nitrogen
Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS*
 COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 9(1-2) 93-100 (2003/2004) Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS* DARIUSZ PLEWCZYNSKI AND LESZEK RYCHLEWSKI BiolnfoBank
COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 9(1-2) 93-100 (2003/2004) Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS* DARIUSZ PLEWCZYNSKI AND LESZEK RYCHLEWSKI BiolnfoBank
Lecture 11: Gene Prediction
 Lecture 11: Gene Prediction Study Chapter 6.11-6.14 1 Gene: A sequence of nucleotides coding for protein Gene Prediction Problem: Determine the beginning and end positions of genes in a genome Where are
Lecture 11: Gene Prediction Study Chapter 6.11-6.14 1 Gene: A sequence of nucleotides coding for protein Gene Prediction Problem: Determine the beginning and end positions of genes in a genome Where are
SUPPLEMENTAL FIGURE LEGENDS. Figure S1: Homology alignment of DDR2 amino acid sequence. Shown are
 SUPPLEMENTAL FIGURE LEGENDS Figure S1: Homology alignment of DDR2 amino acid sequence. Shown are the amino acid sequences of human DDR2, mouse DDR2 and the closest homologs in zebrafish and C. Elegans.
SUPPLEMENTAL FIGURE LEGENDS Figure S1: Homology alignment of DDR2 amino acid sequence. Shown are the amino acid sequences of human DDR2, mouse DDR2 and the closest homologs in zebrafish and C. Elegans.
RNA does not adopt the classic B-DNA helix conformation when it forms a self-complementary double helix
 Reason: RNA has ribose sugar ring, with a hydroxyl group (OH) If RNA in B-from conformation there would be unfavorable steric contact between the hydroxyl group, base, and phosphate backbone. RNA structure
Reason: RNA has ribose sugar ring, with a hydroxyl group (OH) If RNA in B-from conformation there would be unfavorable steric contact between the hydroxyl group, base, and phosphate backbone. RNA structure
Protein Structure Databases, cont. 11/09/05
 11/9/05 Protein Structure Databases (continued) Prediction & Modeling Bioinformatics Seminars Nov 10 Thurs 3:40 Com S Seminar in 223 Atanasoff Computational Epidemiology Armin R. Mikler, Univ. North Texas
11/9/05 Protein Structure Databases (continued) Prediction & Modeling Bioinformatics Seminars Nov 10 Thurs 3:40 Com S Seminar in 223 Atanasoff Computational Epidemiology Armin R. Mikler, Univ. North Texas
DNA stands for deoxyribose nucleic acid
 1 DNA 2 DNA stands for deoxyribose nucleic acid This chemical substance is present in the nucleus of all cells in all living organisms DNA controls all the chemical changes which take place in cells The
1 DNA 2 DNA stands for deoxyribose nucleic acid This chemical substance is present in the nucleus of all cells in all living organisms DNA controls all the chemical changes which take place in cells The
ENZYMES AND METABOLIC PATHWAYS
 ENZYMES AND METABOLIC PATHWAYS This document is licensed under the Attribution-NonCommercial-ShareAlike 2.5 Italy license, available at http://creativecommons.org/licenses/by-nc-sa/2.5/it/ 1. Enzymes build
ENZYMES AND METABOLIC PATHWAYS This document is licensed under the Attribution-NonCommercial-ShareAlike 2.5 Italy license, available at http://creativecommons.org/licenses/by-nc-sa/2.5/it/ 1. Enzymes build
Università degli Studi di Roma La Sapienza
 Università degli Studi di Roma La Sapienza. Tutore Prof.ssa Anna Tramontano Coordinatore Prof. Marco Tripodi Dottorando Daniel Carbajo Pedrosa Dottorato di Ricerca in Scienze Pasteuriane XXIV CICLO Docente
Università degli Studi di Roma La Sapienza. Tutore Prof.ssa Anna Tramontano Coordinatore Prof. Marco Tripodi Dottorando Daniel Carbajo Pedrosa Dottorato di Ricerca in Scienze Pasteuriane XXIV CICLO Docente
1. DNA replication. (a) Why is DNA replication an essential process?
 Why is DNA replication an essential process?") ame Section 7.014 Problem Set 3 Please print out this problem set and record your answers on the printed copy. Answers to this problem set are to be turned in to the box outside 68120 by 5:00pm on Friday
ame Section 7.014 Problem Set 3 Please print out this problem set and record your answers on the printed copy. Answers to this problem set are to be turned in to the box outside 68120 by 5:00pm on Friday
Ch 10 Molecular Biology of the Gene
 Ch 10 Molecular Biology of the Gene For Next Week Lab -Hand in questions from 4 and 5 by TUES in my mailbox (Biology Office) -Do questions for Lab 6 for next week -Lab practical next week Lecture Read
Ch 10 Molecular Biology of the Gene For Next Week Lab -Hand in questions from 4 and 5 by TUES in my mailbox (Biology Office) -Do questions for Lab 6 for next week -Lab practical next week Lecture Read
Amino Acid Distribution Rules Predict Protein Fold: Protein Grammar for Beta-Strand Sandwich-Like Structures
 Biomolecules 2015, 5, 41-59; doi:10.3390/biom5010041 Article OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Amino Acid Distribution Rules Predict Protein Fold: Protein Grammar
Biomolecules 2015, 5, 41-59; doi:10.3390/biom5010041 Article OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Amino Acid Distribution Rules Predict Protein Fold: Protein Grammar
Cristian Micheletti SISSA (Trieste)
") Cristian Micheletti SISSA (Trieste) michelet@sissa.it Mar 2009 5pal - parvalbumin Calcium-binding protein HEADER CALCIUM-BINDING PROTEIN 25-SEP-91 5PAL 5PAL 2 COMPND PARVALBUMIN (ALPHA LINEAGE) 5PAL 3
Cristian Micheletti SISSA (Trieste) michelet@sissa.it Mar 2009 5pal - parvalbumin Calcium-binding protein HEADER CALCIUM-BINDING PROTEIN 25-SEP-91 5PAL 5PAL 2 COMPND PARVALBUMIN (ALPHA LINEAGE) 5PAL 3
Introduction to Bioinformatics
 Introduction to Bioinformatics Changhui (Charles) Yan Old Main 401 F http://www.cs.usu.edu www.cs.usu.edu/~cyan 1 How Old Is The Discipline? "The term bioinformatics is a relatively recent invention, not
Introduction to Bioinformatics Changhui (Charles) Yan Old Main 401 F http://www.cs.usu.edu www.cs.usu.edu/~cyan 1 How Old Is The Discipline? "The term bioinformatics is a relatively recent invention, not
Proteins the primary biological macromolecules of living organisms
 Proteins the primary biological macromolecules of living organisms Protein structure and folding Primary Secondary Tertiary Quaternary structure of proteins Structure of Proteins Protein molecules adopt
Proteins the primary biological macromolecules of living organisms Protein structure and folding Primary Secondary Tertiary Quaternary structure of proteins Structure of Proteins Protein molecules adopt
Disease and selection in the human genome 3
 Disease and selection in the human genome 3 Ka/Ks revisited Please sit in row K or forward RBFD: human populations, adaptation and immunity Neandertal Museum, Mettman Germany Sequence genome Measure expression
Disease and selection in the human genome 3 Ka/Ks revisited Please sit in row K or forward RBFD: human populations, adaptation and immunity Neandertal Museum, Mettman Germany Sequence genome Measure expression
Homology Modeling of Mouse orphan G-protein coupled receptors Q99MX9 and G2A
 Quality in Primary Care (2016) 24 (2): 49-57 2016 Insight Medical Publishing Group Research Article Homology Modeling of Mouse orphan G-protein Research Article Open Access coupled receptors Q99MX9 and
Quality in Primary Care (2016) 24 (2): 49-57 2016 Insight Medical Publishing Group Research Article Homology Modeling of Mouse orphan G-protein Research Article Open Access coupled receptors Q99MX9 and
BLAST. compared with database sequences Sequences with many matches to high- scoring words are used for final alignments
 BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
Comparative Bioinformatics. BSCI348S Fall 2003 Midterm 1
 BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
Genome Sequence Assembly
 Genome Sequence Assembly Learning Goals: Introduce the field of bioinformatics Familiarize the student with performing sequence alignments Understand the assembly process in genome sequencing Introduction:
Genome Sequence Assembly Learning Goals: Introduce the field of bioinformatics Familiarize the student with performing sequence alignments Understand the assembly process in genome sequencing Introduction:
Homology modeling and structural validation of tissue factor pathway inhibitor
 www.bioinformation.net Hypothesis Volume 9(16) Homology modeling and structural validation of tissue factor pathway inhibitor Piyush Agrawal, Zoozeal Thakur & Mahesh Kulharia* Centre for Bioinformatics,
www.bioinformation.net Hypothesis Volume 9(16) Homology modeling and structural validation of tissue factor pathway inhibitor Piyush Agrawal, Zoozeal Thakur & Mahesh Kulharia* Centre for Bioinformatics,
Basic Biology. Gina Cannarozzi. 28th October Basic Biology. Gina. Introduction DNA. Proteins. Central Dogma.
 Cannarozzi 28th October 2005 Class Overview RNA Protein Genomics Transcriptomics Proteomics Genome wide Genome Comparison Microarrays Orthology: Families comparison and Sequencing of Transcription factor
Cannarozzi 28th October 2005 Class Overview RNA Protein Genomics Transcriptomics Proteomics Genome wide Genome Comparison Microarrays Orthology: Families comparison and Sequencing of Transcription factor
Retrieving and Viewing Protein Structures from the Protein Data Base
 Retrieving and Viewing Protein Structures from the Protein Data Base 7.88J Protein Folding Prof. David Gossard September 15, 2003 1 PDB Acknowledgements The Protein Data Bank (PDB - http://www.pdb.org/)
Retrieving and Viewing Protein Structures from the Protein Data Base 7.88J Protein Folding Prof. David Gossard September 15, 2003 1 PDB Acknowledgements The Protein Data Bank (PDB - http://www.pdb.org/)
What s New in Discovery Studio 2.5.5
 What s New in Discovery Studio 2.5.5 Discovery Studio takes modeling and simulations to the next level. It brings together the power of validated science on a customizable platform for drug discovery research.
What s New in Discovery Studio 2.5.5 Discovery Studio takes modeling and simulations to the next level. It brings together the power of validated science on a customizable platform for drug discovery research.
36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L-
 36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L- 37. The essential fatty acids are A. palmitic acid B. linoleic acid C. linolenic
36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L- 37. The essential fatty acids are A. palmitic acid B. linoleic acid C. linolenic
CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS
 1 CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS * Some contents are adapted from Dr. Jean Gao at UT Arlington Mingon Kang, PhD Computer Science, Kennesaw State University 2 Genetics The discovery of
1 CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS * Some contents are adapted from Dr. Jean Gao at UT Arlington Mingon Kang, PhD Computer Science, Kennesaw State University 2 Genetics The discovery of
1. DNA, RNA structure. 2. DNA replication. 3. Transcription, translation
 1. DNA, RNA structure 2. DNA replication 3. Transcription, translation DNA and RNA are polymers of nucleotides DNA is a nucleic acid, made of long chains of nucleotides Nucleotide Phosphate group Nitrogenous
1. DNA, RNA structure 2. DNA replication 3. Transcription, translation DNA and RNA are polymers of nucleotides DNA is a nucleic acid, made of long chains of nucleotides Nucleotide Phosphate group Nitrogenous
APPENDIX. Appendix. Table of Contents. Ethics Background. Creating Discussion Ground Rules. Amino Acid Abbreviations and Chemistry Resources
 Appendix Table of Contents A2 A3 A4 A5 A6 A7 A9 Ethics Background Creating Discussion Ground Rules Amino Acid Abbreviations and Chemistry Resources Codons and Amino Acid Chemistry Behind the Scenes with
Appendix Table of Contents A2 A3 A4 A5 A6 A7 A9 Ethics Background Creating Discussion Ground Rules Amino Acid Abbreviations and Chemistry Resources Codons and Amino Acid Chemistry Behind the Scenes with
Introduction to Bioinformatics CPSC 265. What is bioinformatics? Textbooks
 Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
ONLINE BIOINFORMATICS RESOURCES
 Dedan Githae Email: d.githae@cgiar.org BecA-ILRI Hub; Nairobi, Kenya 16 May, 2014 ONLINE BIOINFORMATICS RESOURCES Introduction to Molecular Biology and Bioinformatics (IMBB) 2014 The larger picture.. Lower
Dedan Githae Email: d.githae@cgiar.org BecA-ILRI Hub; Nairobi, Kenya 16 May, 2014 ONLINE BIOINFORMATICS RESOURCES Introduction to Molecular Biology and Bioinformatics (IMBB) 2014 The larger picture.. Lower
Insight II. Homology. March Scranton Road San Diego, CA / Fax: 858/
 Insight II Homology March 2000 9685 Scranton Road San Diego, CA 92121-3752 858/458-9990 Fax: 858/458-0136 Copyright * This document is copyright 2000, Molecular Simulations Inc., a subsidiary of Pharmacopeia,
Insight II Homology March 2000 9685 Scranton Road San Diego, CA 92121-3752 858/458-9990 Fax: 858/458-0136 Copyright * This document is copyright 2000, Molecular Simulations Inc., a subsidiary of Pharmacopeia,
ELE4120 Bioinformatics. Tutorial 5
 ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
Chimp Sequence Annotation: Region 2_3
 Chimp Sequence Annotation: Region 2_3 Jeff Howenstein March 30, 2007 BIO434W Genomics 1 Introduction We received region 2_3 of the ChimpChunk sequence, and the first step we performed was to run RepeatMasker
Chimp Sequence Annotation: Region 2_3 Jeff Howenstein March 30, 2007 BIO434W Genomics 1 Introduction We received region 2_3 of the ChimpChunk sequence, and the first step we performed was to run RepeatMasker
Protein Structure Prediction by Constraint Logic Programming
 MPRI C2-19 Protein Structure Prediction by Constraint Logic Programming François Fages, Constraint Programming Group, INRIA Rocquencourt mailto:francois.fages@inria.fr http://contraintes.inria.fr/ Molecules
MPRI C2-19 Protein Structure Prediction by Constraint Logic Programming François Fages, Constraint Programming Group, INRIA Rocquencourt mailto:francois.fages@inria.fr http://contraintes.inria.fr/ Molecules
Sequence Based Function Annotation. Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University
 Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
Bi 8 Lecture 7. Ellen Rothenberg 26 January Reading: Ch. 3, pp ; panel 3-1
 Bi 8 Lecture 7 PROTEIN STRUCTURE, Functional analysis, and evolution Ellen Rothenberg 26 January 2016 Reading: Ch. 3, pp. 109-134; panel 3-1 (end with free amine) aromatic, hydrophobic small, hydrophilic
Bi 8 Lecture 7 PROTEIN STRUCTURE, Functional analysis, and evolution Ellen Rothenberg 26 January 2016 Reading: Ch. 3, pp. 109-134; panel 3-1 (end with free amine) aromatic, hydrophobic small, hydrophilic
Name Date of Data Collection. Class Period Lab Days/Period Teacher
 Comparing Primates (adapted from Comparing Primates Lab, page 431-438, Biology Lab Manual, by Miller and Levine, Prentice Hall Publishers, copyright 2000, ISBN 0-13-436796-0) Background: One of the most
Comparing Primates (adapted from Comparing Primates Lab, page 431-438, Biology Lab Manual, by Miller and Levine, Prentice Hall Publishers, copyright 2000, ISBN 0-13-436796-0) Background: One of the most
7.014 Quiz II Handout
 7.014 Quiz II Handout Quiz II: Wednesday, March 17 12:05-12:55 54-100 **This will be a closed book exam** Quiz Review Session: Friday, March 12 7:00-9:00 pm room 54-100 Open Tutoring Session: Tuesday,
7.014 Quiz II Handout Quiz II: Wednesday, March 17 12:05-12:55 54-100 **This will be a closed book exam** Quiz Review Session: Friday, March 12 7:00-9:00 pm room 54-100 Open Tutoring Session: Tuesday,
Supplementary Table 1: List of CH3 domain interface residues in the first chain (A) and
 and") Supplementary Tables Supplementary Table 1: List of CH3 domain interface residues in the first chain (A) and their side chain contacting residues in the second chain (B) a Interface Res. in Contacting
Supplementary Tables Supplementary Table 1: List of CH3 domain interface residues in the first chain (A) and their side chain contacting residues in the second chain (B) a Interface Res. in Contacting
BIL 256 Cell and Molecular Biology Lab Spring, 2007 BACKGROUND INFORMATION I. PROTEIN COMPOSITION AND STRUCTURE: A REVIEW OF THE BASICS
 BIL 256 Cell and Molecular Biology Lab Spring, 2007 BACKGROUND INFORMATION I. PROTEIN COMPOSITION AND STRUCTURE: A REVIEW OF THE BASICS Proteins occupy a central position in the structure and function
BIL 256 Cell and Molecular Biology Lab Spring, 2007 BACKGROUND INFORMATION I. PROTEIN COMPOSITION AND STRUCTURE: A REVIEW OF THE BASICS Proteins occupy a central position in the structure and function
Supplementary Materials. Figure S1 Chemical structures of cisplatin and carboplatin.
 Supplementary Materials Figure S1 Chemical structures of cisplatin and carboplatin. Figure S2 (a) RMS difference plot between the RT and 100K structures for the carboplatin_dmso case after 13 months of
Supplementary Materials Figure S1 Chemical structures of cisplatin and carboplatin. Figure S2 (a) RMS difference plot between the RT and 100K structures for the carboplatin_dmso case after 13 months of
Introduction to Bioinformatics
 Introduction to Bioinformatics http://1.51.212.243/bioinfo.html Dr. rer. nat. Jing Gong Cancer Research Center School of Medicine, Shandong University 2011.10.19 1 Chapter 4 Structure 2 Protein Structure
Introduction to Bioinformatics http://1.51.212.243/bioinfo.html Dr. rer. nat. Jing Gong Cancer Research Center School of Medicine, Shandong University 2011.10.19 1 Chapter 4 Structure 2 Protein Structure
Biomolecules: lecture 6
 Biomolecules: lecture 6 - to learn the basics on how DNA serves to make RNA = transcription - to learn how the genetic code instructs protein synthesis - to learn the basics on how proteins are synthesized
Biomolecules: lecture 6 - to learn the basics on how DNA serves to make RNA = transcription - to learn how the genetic code instructs protein synthesis - to learn the basics on how proteins are synthesized
Proteins Higher Order Structures
 Proteins Higher Order Structures Dr. Mohammad Alsenaidy Department of Pharmaceutics College of Pharmacy King Saud University Office: AA 101 msenaidy@ksu.edu.sa Previously on PHT 426!! Protein Structures
Proteins Higher Order Structures Dr. Mohammad Alsenaidy Department of Pharmaceutics College of Pharmacy King Saud University Office: AA 101 msenaidy@ksu.edu.sa Previously on PHT 426!! Protein Structures
BIRKBECK COLLEGE (University of London)
") BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
MOLEBIO LAB #3: Electrophoretic Separation of Proteins
 MOLEBIO LAB #3: Electrophoretic Separation of Proteins Introduction: Proteins occupy a central position in the structure and function of all living organisms. Some proteins serve as structural components
MOLEBIO LAB #3: Electrophoretic Separation of Proteins Introduction: Proteins occupy a central position in the structure and function of all living organisms. Some proteins serve as structural components
Introduction to Molecular Biology
 Introduction to Molecular Biology Content Cells and organisms Molecules of life (Biomolecules) Central dogma of molecular biology Genes and gene expression @: Most pictures have been freely obtained from:
Introduction to Molecular Biology Content Cells and organisms Molecules of life (Biomolecules) Central dogma of molecular biology Genes and gene expression @: Most pictures have been freely obtained from:
Gene Identification in silico
 Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Genetic Algorithms For Protein Threading
 From: ISMB-98 Proceedings. Copyright 1998, AAAI (www.aaai.org). All rights reserved. Genetic Algorithms For Protein Threading Jacqueline Yadgari #, Amihood Amir #, Ron Unger* # Department of Mathematics
From: ISMB-98 Proceedings. Copyright 1998, AAAI (www.aaai.org). All rights reserved. Genetic Algorithms For Protein Threading Jacqueline Yadgari #, Amihood Amir #, Ron Unger* # Department of Mathematics
The study of protein secondary structure and stability at equilibrium ABSTRACT
 The study of protein secondary structure and stability at equilibrium Michelle Planicka Dept. of Physics, North Georgia College and State University, Dahlonega, GA REU, Dept. of Physics, University of
The study of protein secondary structure and stability at equilibrium Michelle Planicka Dept. of Physics, North Georgia College and State University, Dahlonega, GA REU, Dept. of Physics, University of
Daily Agenda. Warm Up: Review. Translation Notes Protein Synthesis Practice. Redos
 Daily Agenda Warm Up: Review Translation Notes Protein Synthesis Practice Redos 1. What is DNA Replication? 2. Where does DNA Replication take place? 3. Replicate this strand of DNA into complimentary
Daily Agenda Warm Up: Review Translation Notes Protein Synthesis Practice Redos 1. What is DNA Replication? 2. Where does DNA Replication take place? 3. Replicate this strand of DNA into complimentary
STRUCTURAL BIOLOGY. α/β structures Closed barrels Open twisted sheets Horseshoe folds
 STRUCTURAL BIOLOGY α/β structures Closed barrels Open twisted sheets Horseshoe folds The α/β domains Most frequent domain structures are α/β domains: A central parallel or mixed β sheet Surrounded by α
STRUCTURAL BIOLOGY α/β structures Closed barrels Open twisted sheets Horseshoe folds The α/β domains Most frequent domain structures are α/β domains: A central parallel or mixed β sheet Surrounded by α
Introduction to Bioinformatics
 Introduction to Bioinformatics Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com
Introduction to Bioinformatics Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com
Main-Chain Conformational Tendencies of Amino Acids
 PROTEINS: Structure, Function, and Bioinformatics 60:679 689 (2005) Main-Chain Conformational Tendencies of Amino Acids Robert J. Anderson, 1,2 Zhiping Weng, 2 Robert K. Campbell, 1 and Xuliang Jiang 1
PROTEINS: Structure, Function, and Bioinformatics 60:679 689 (2005) Main-Chain Conformational Tendencies of Amino Acids Robert J. Anderson, 1,2 Zhiping Weng, 2 Robert K. Campbell, 1 and Xuliang Jiang 1